We are pleased to share a selection of our referee-recommended HOT articles for July. We hope you enjoy reading these articles and congratulations to all the authors whose articles are featured! As always, Chemical Science is free to read & download. You can find our full 2020 HOT article collection here.
Exohedral functionalization vs. core expansion of siliconoids with Group 9 metals: catalytic activity in alkene isomerization
Nadine E. Poitiers, Luisa Giarrana, Volker Huch, Michael Zimmer and David Scheschkewitz
Chem. Sci., 2020, Advance Article
DOI: 10.1039/D0SC02861D
Deoxygenative α-alkylation and α-arylation of 1,2-dicarbonyls
Shengfei Jin, Hang T. Dang, Graham C. Haug, Viet D. Nguyen, Hadi D. Arman and Oleg V. Larionov
Chem. Sci., 2020, Advance Article
DOI: 10.1039/D0SC03118F
Cellular uptake and targeting of low dispersity, dual emissive, segmented block copolymer nanofibers
Steven T. G. Street, Yunxiang He, Xu-Hui Jin, Lorna Hodgson, Paul Verkade and Ian Manners
Chem. Sci., 2020, Advance Article
DOI: 10.1039/D0SC02593C
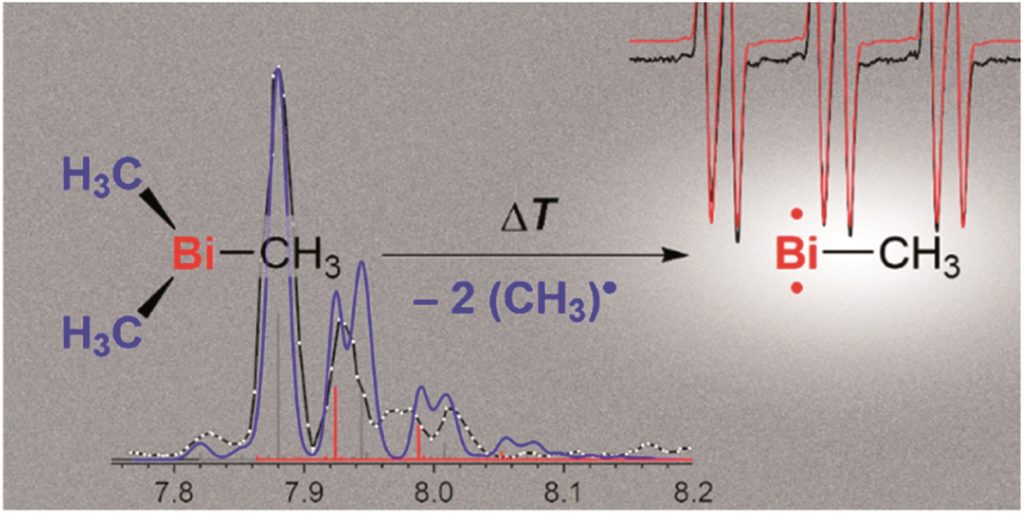
Mechanochemical synthesis of glycine oligomers in a virtual rotational diamond anvil cell
Brad A. Steele, Nir Goldman, I-Feng W. Kuo and Matthew P. Kroonblawd
Chem. Sci., 2020, Advance Article
DOI: 10.1039/D0SC00755B
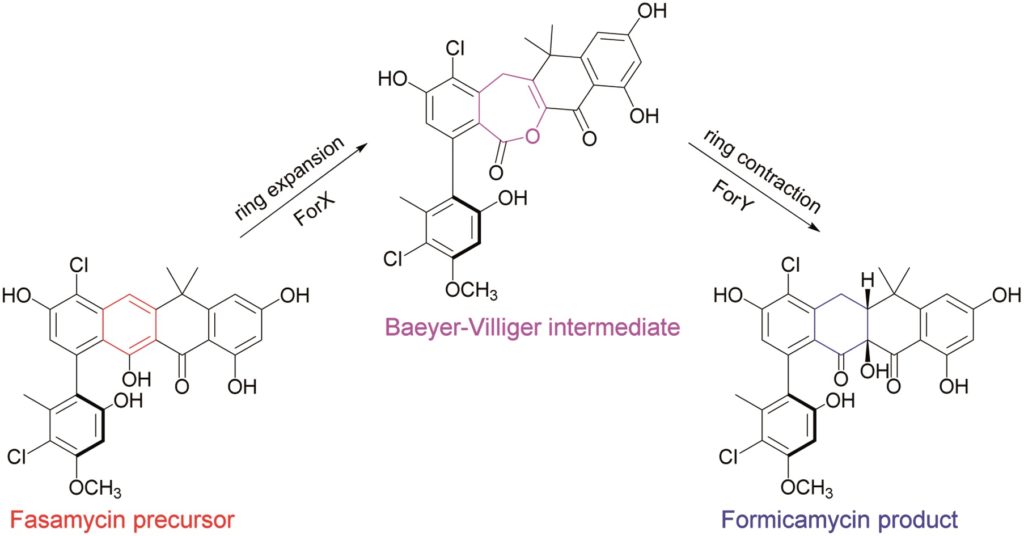
Total synthesis of endiandric acid J and beilcyclone A from cyclooctatetraene
Oussama Yahiaoui, Adrian Almass and Thomas Fallon
Chem. Sci., 2020, Advance Article
DOI: 10.1039/D0SC03073B
Template effects of vesicles in dynamic covalent chemistry
Carlo Bravin and Christopher A. Hunter
Chem. Sci., 2020, Advance Article
DOI: 10.1039/D0SC03185B
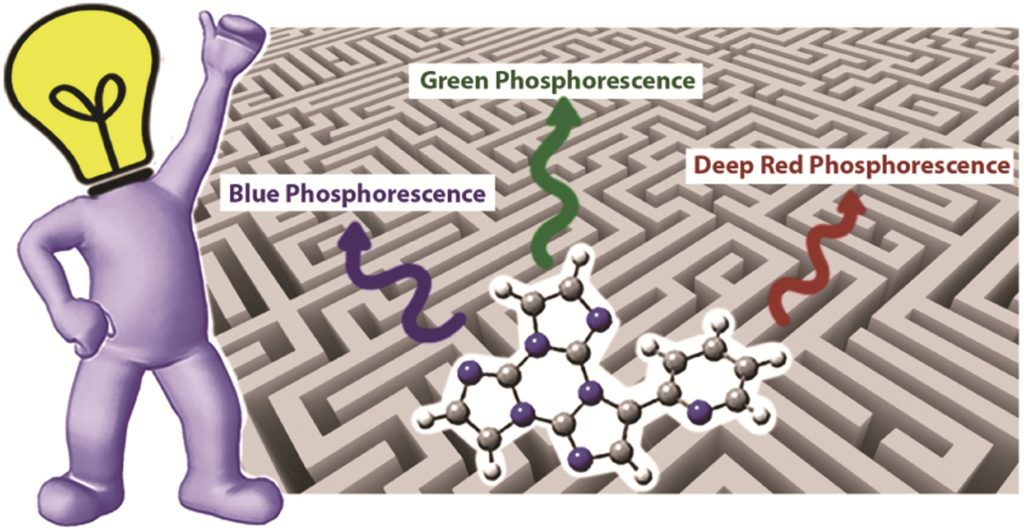
Simultaneously boosting the conjugation, brightness and solubility of organic fluorophores by using AIEgens
Ji Qi, Xingchen Duan, Yuanjing Cai, Shaorui Jia, Chao Chen, Zheng Zhao, Ying Li, Hui-Qing Peng, Ryan T. K. Kwok, Jacky W. Y. Lam, Dan Ding and Ben Zhong Tang
Chem. Sci., 2020, Advance Article
DOI: 10.1039/D0SC03423A
Enhanced enzymatic activity exerted by a packed assembly of a single type of enzyme
Huyen Dinh, Eiji Nakata, Kaori Mutsuda-Zapater, Masayuki Saimura, Masahiro Kinoshita and Takashi Morii
Chem. Sci., 2020, Advance Article
DOI: 10.1039/D0SC03498C
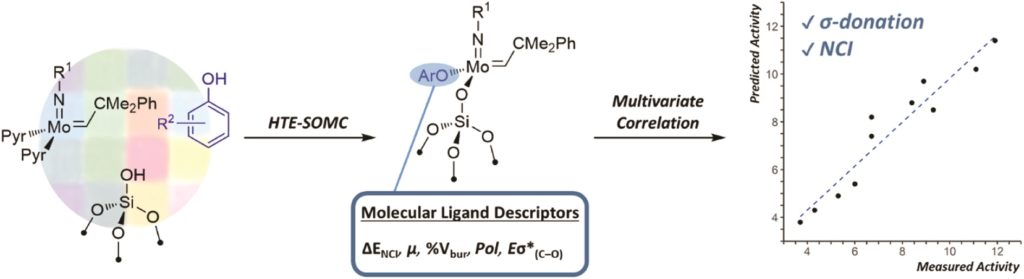
Structure-activity relationships in well-defined conjugated oligomer photocatalysts for hydrogen production from water
Catherine M. Aitchison, Michael Sachs, Marc Little, Liam Wilbraham, Nick J. Brownbill, Chris Kane, Frédéric Blanc, Martijn Zwijnenburg, James Durrant, Reiner Sebastian Sprick and Andrew Cooper
Chem. Sci., 2020, Accepted Manuscript
DOI: 10.1039/D0SC02675A

Submit to Chemical Science today! Check out our author guidelines for information on our article types or find out more about the advantages of publishing in a Royal Society of Chemistry journal.
Keep up to date with our latest articles, reviews, collections & more by following us on Twitter. You can also keep informed by signing up to our E-Alerts.








")




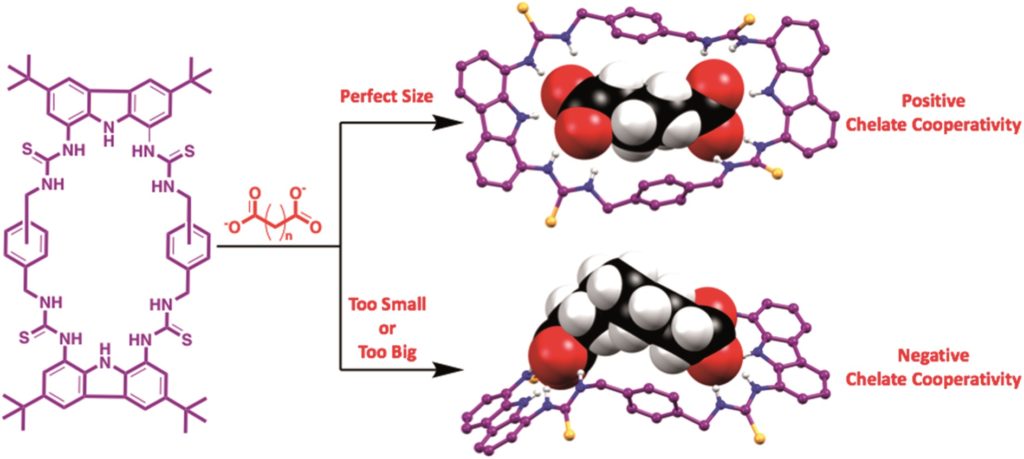
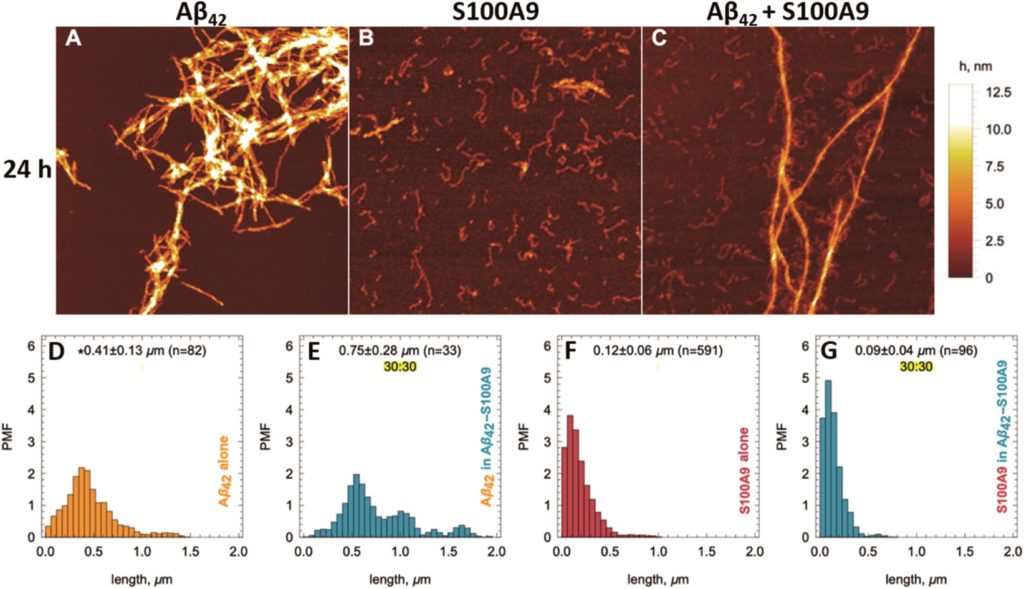

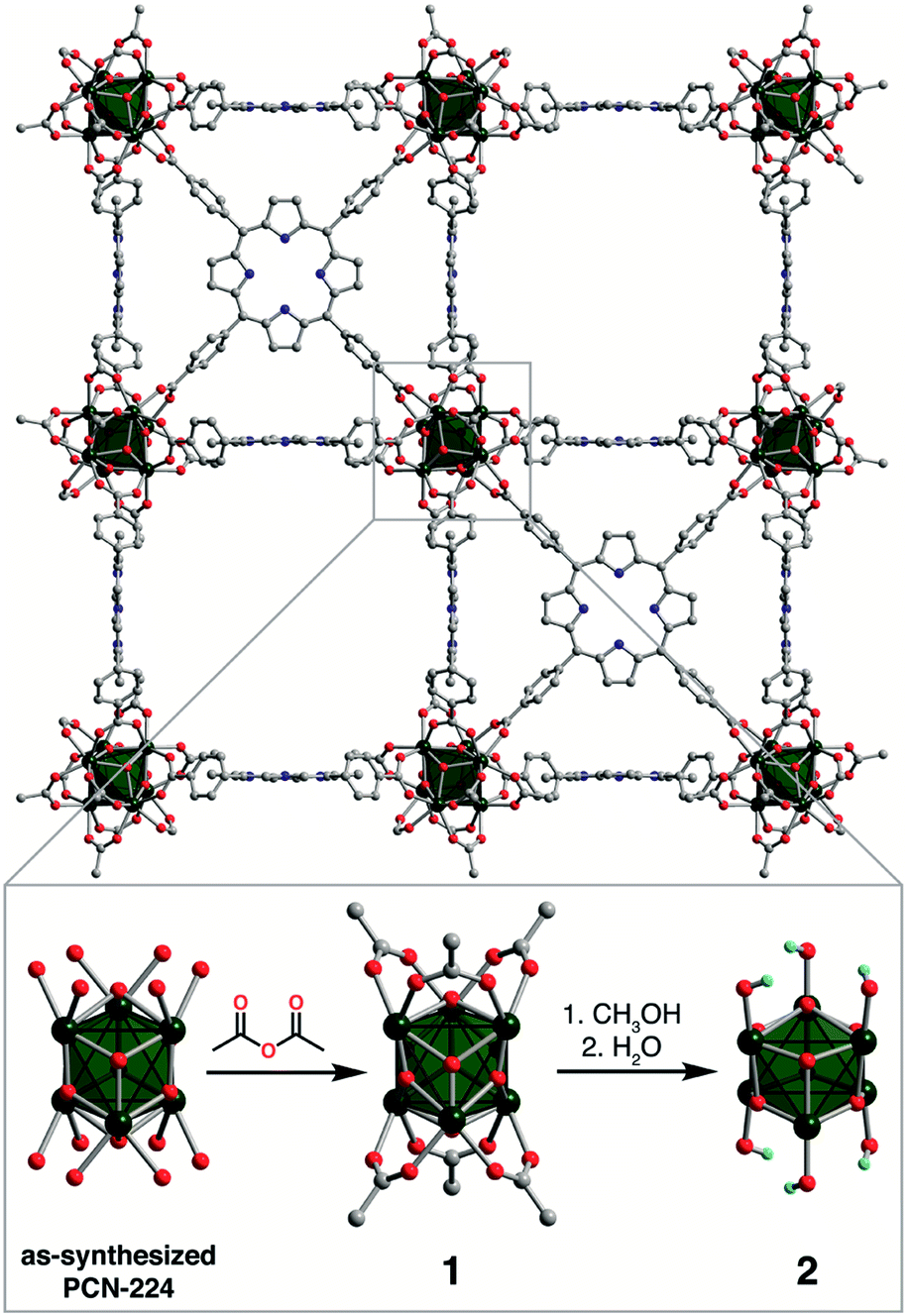
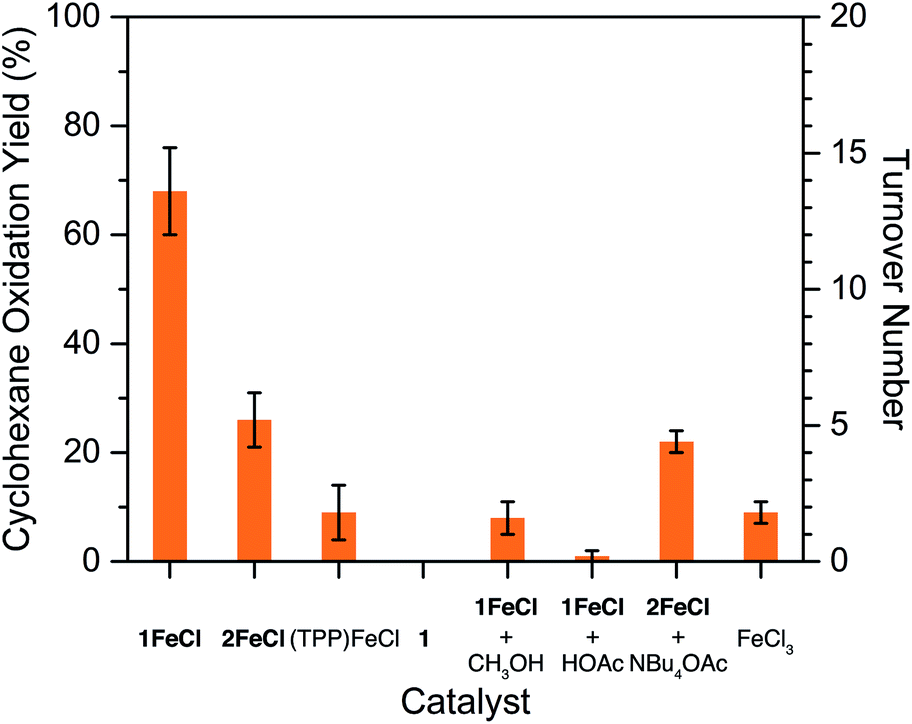
 Tianyu Liu obtained his Ph.D. (2017) in Chemistry from the University of California, Santa Cruz, in the United States. He is passionate about the communication of scientific endeavors to both the general public and other scientists with diverse research expertise to introduce cutting-edge research to broad audiences. He is a blog writer for Chem. Comm. and Chem. Sci. More information about him can be found at
Tianyu Liu obtained his Ph.D. (2017) in Chemistry from the University of California, Santa Cruz, in the United States. He is passionate about the communication of scientific endeavors to both the general public and other scientists with diverse research expertise to introduce cutting-edge research to broad audiences. He is a blog writer for Chem. Comm. and Chem. Sci. More information about him can be found at 