Recent decades have established that metal-organic frameworks (MOFs) are a pretty cool class of materials, with potential applications across a range of fields. In particular, their high porosities make them extremely attractive for storing a variety of gases, including possible fuels like methane and hydrogen. Two primary strategies have emerged to store H2 and CH4 in MOFs – synthesizing materials with unsaturated metals that can strongly bind to the target and synthesizing materials with small pores where multiple weak interactions combine to produce strong binding. Of course, these MOFs are designed with a single specific target in mind, making them synthetically complex and useful for storing only one type of molecule. Ideally, new MOFs with relatively straightforward syntheses that can bind multiple targets could be developed.
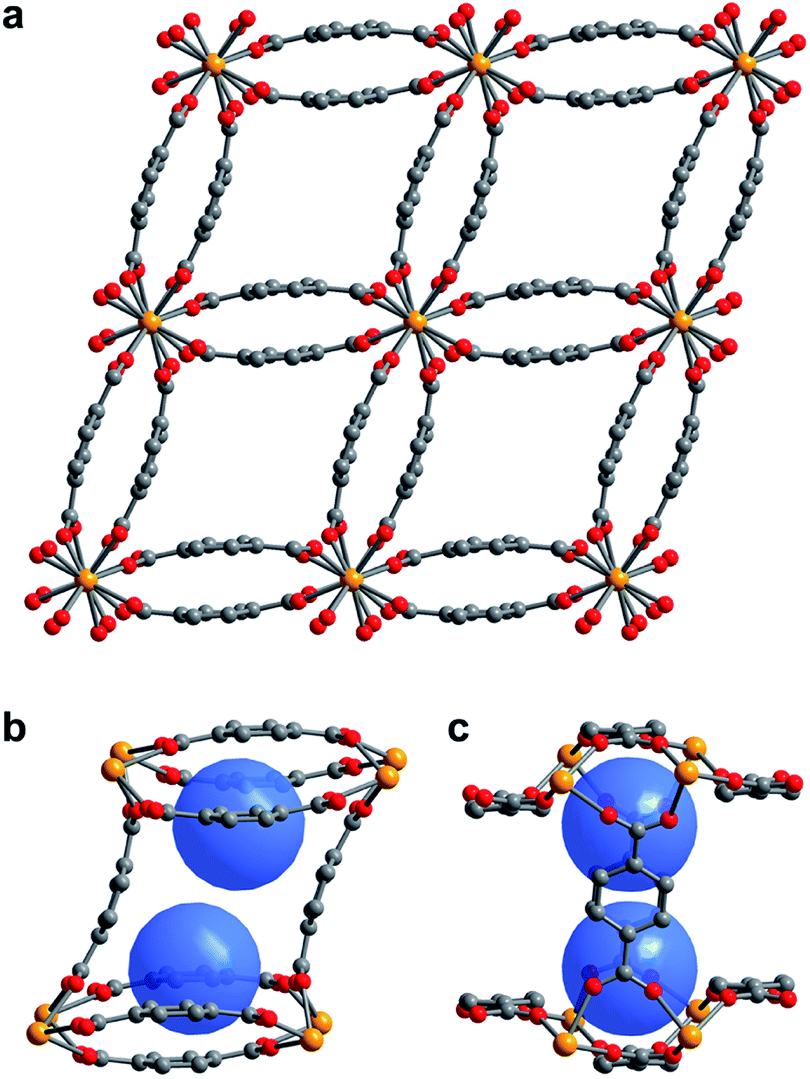
Figure 1. (a) Single crystal X-ray diffraction structure of 1–H2O. (b,c) View of the pores of 1–H2O showing binding pocket.
Scientists in the United States took a hybrid approach, creating MOFs with small, but flexible binding pockets. While the concept feels relatively straightforward and intuitive, it’s of course more complicated in practice. The MOF needs to hit a Goldilocks zone in terms of flexibility, where only a small number of select targets will bind rather than a wide range of gases. The researchers accomplished this by using an actinide (depleted uranium) as the metal nodes for their MOF as its tendency to adopt high coordination numbers should result in smaller pockets and limit possible rearrangements of the flexible linker. Also, the descriptor “as it is only mildly radioactive” is something I hadn’t read about a material before and rather caught my attention. The crystalline material, referred to as 1-H2O (Figure 1), was straightforwardly synthesized in an autoclave and isolated in relatively high yields. It features pores with two pockets that are capped by the bowl-shaped linkers. As synthesized, the pockets are occupied by water molecules that can be removed by heating the MOF under a dynamic vacuum.
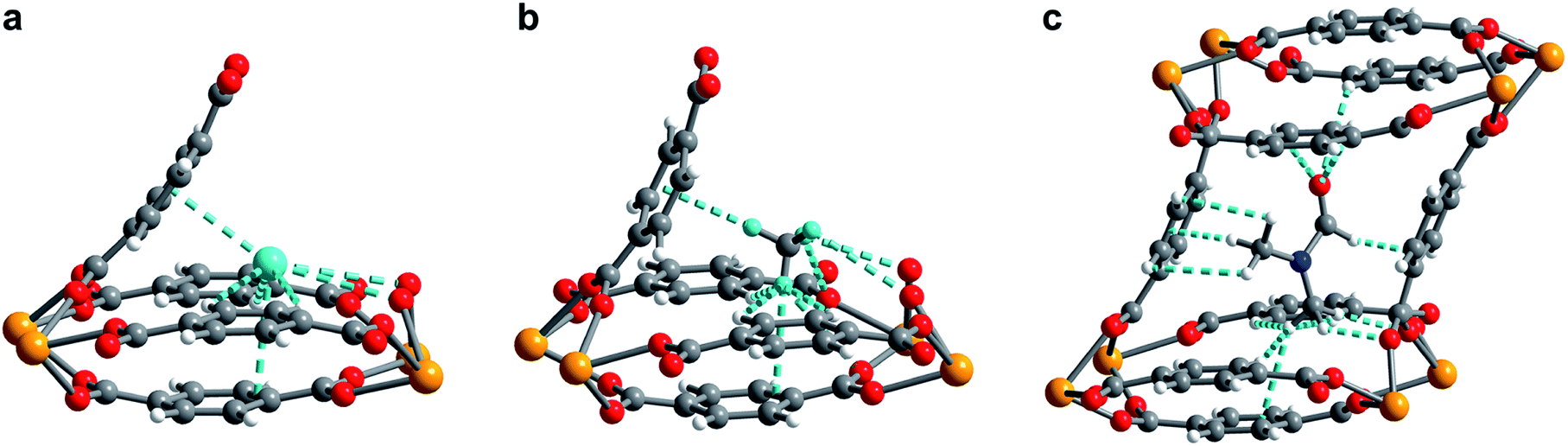
Figure 2. (a) Neutron powder diffraction structure of D2 adsorbed at site I in 1–D2. (b) Neutron powder diffraction structure of CD4 adsorbed at site I in 1–CD4. (c) Powder X-ray diffraction structure of DMF adsorbed inside the pore of U(bdc)2.
The MOF maintains its structural integrity after water removal, actually expanding slightly. This indicates that the MOF will contract upon binding, locking the target into place in the pocket. The researchers found that the MOF rapidly uptakes both H2 and CH4 at low temperatures, but the precise nature of the binding pocket adjustments can’t be determined by gas adsorption studies. To probe the structural details, the researchers turned to neutron powder diffraction to probe the binding of deuterated molecules to the MOF (Figure 2). The obtained structures show clear, cooperative effects that cause the adjustments to the binding pocket. The multiple different interactions allow the flexible structure to fit the two different adsorbates of interest, binding them both strongly. This work demonstrates the utility and versatility of flexible MOFs for adsorbing different gases with design principles that should be transferrable to non-radioactive materials.
To find out more, please read:
Dominik P. Halter, Ryan A. Klein, Michael A. Boreen, Benjamin A. Trump, Craig M. Brown and Jeffrey R. Long
Chem. Sci., 2020, Advance Article
About the blogger:
 Dr. Beth Mundy is a recent PhD in chemistry from the Cossairt lab at the University of Washington in Seattle, Washington. Her research focused on developing new and better ways to synthesize nanomaterials for energy applications. She is often spotted knitting in seminars or with her nose in a good book. You can find her on Twitter at @BethMundySci.
Dr. Beth Mundy is a recent PhD in chemistry from the Cossairt lab at the University of Washington in Seattle, Washington. Her research focused on developing new and better ways to synthesize nanomaterials for energy applications. She is often spotted knitting in seminars or with her nose in a good book. You can find her on Twitter at @BethMundySci.








")