We are delighted to introduce our two newest Editorial Board members for Chemical Science – Professor Nobuhiro Yanai and Professor Jun Cheng. Both will be handling manuscripts submitted to the journal in their roles as Associate Editors.
Professor Nobuhiro Yanai 
Nobuhiro Yanai is a Professor in the Department of Chemistry at the University of Tokyo, having previously been an Associate Professor at Kyushu University. His research covers a broad range of materials science and photochemistry topics, with a particular focus on quantum sensing and control, dynamic nuclear polarisation and photon upconversion.
“I am delighted and honoured to join Chemical Science as an Associate Editor. Chemical Science has long been a home for pioneering and interdisciplinary research that expands the boundaries of chemistry and connects diverse scientific disciplines. I look forward to working with authors and reviewers from around the world to help communicate the most exciting advances in our field. I am particularly excited by emerging opportunities in photochemistry, spin chemistry, magnetic resonance, and molecular quantum technologies, and I welcome submissions that combine fundamental discovery with broad scientific impact and open new directions in chemistry.”
Discover some of the topics that Nobuhiro will be considering in our Most Popular Photochemistry and Physical Chemistry collections and read through his recent Chemical Science publications below:
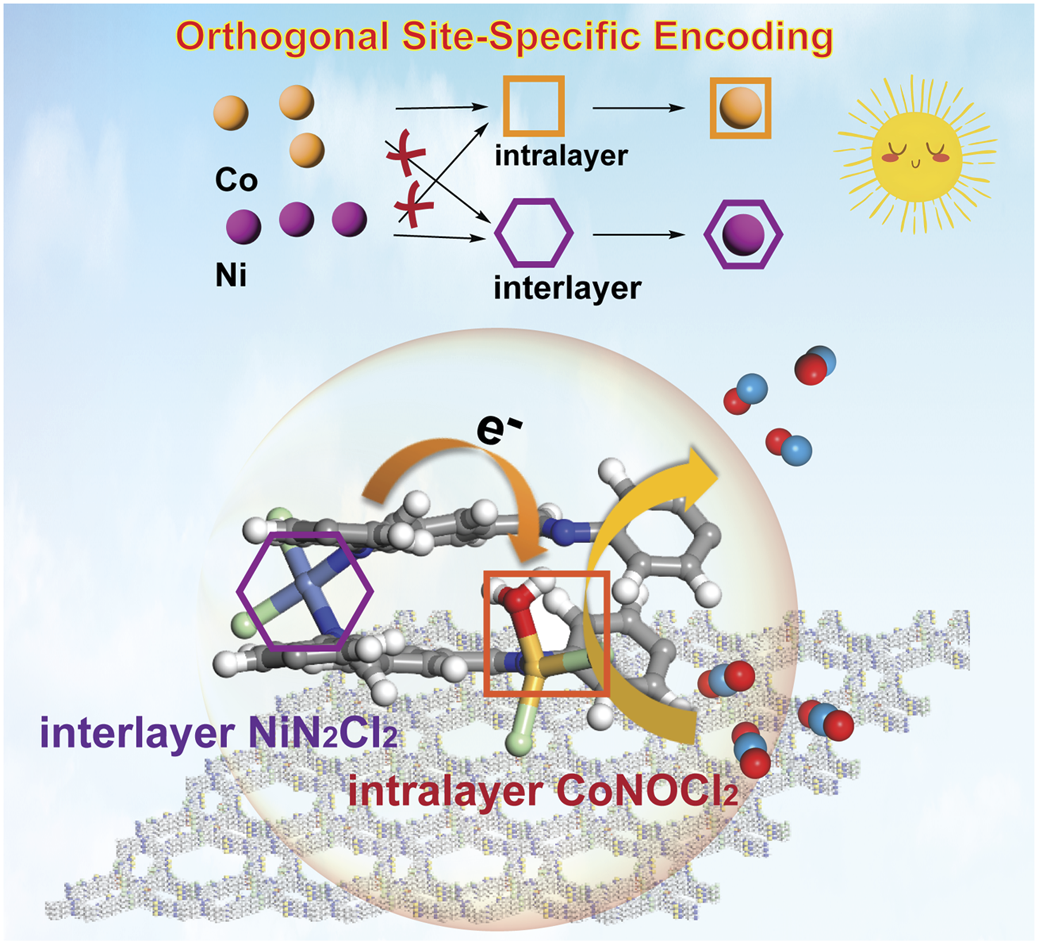
Crystalline organic monoliths with bicontinuous porosity Nobuhiro Yanai et al. Chem. Sci., 2024, 15, 11500-11506
Enhancing the statistical probability factor in triplet–triplet annihilation photon upconversion via TIPS functionalization Pankaj Bharmoria, Kasper Moth-Poulsen et al. Chem. Sci., 2025, 16, 20255-20264.
Professor Jun Cheng
Jun Cheng is a Professor at Xiamen University, having previously held positions at the University of Cambridge and the University of Aberdeen. His research and expertise covers a broad range of topics in computational chemistry, machine learning, electrochemistry and catalysis.
“It is a great honour to join the editorial team of the RSC’s flagship journal, Chemical Science. I am truly excited by the opportunity to work with such an outstanding group of editors and to contribute to the journal’s mission of publishing high-quality research that advances the field of chemistry. I look forward to collaborating with the editorial team to support the dissemination of excellent scientific work, particularly in areas closely related to my own research interests, including computational chemistry, artificial intelligence for chemistry, electrochemistry, and catalysis. These fields are evolving rapidly and are playing an increasingly important role in addressing fundamental scientific questions as well as major challenges in energy, materials, and sustainability. As a member of the editorial team, I hope to help identify, evaluate, and promote rigorous, innovative, and impactful manuscripts. I am especially enthusiastic about supporting interdisciplinary studies that connect chemical theory, simulation, data-driven methods, and experimental insights. I believe that the journal will continue to serve as an important platform for shaping the future of chemical research, and I am delighted to contribute to this effort.”
Check out some of the research fields Jun will be considering work on in our Most Popular Theoretical and Computational Chemistry and Catalysis collections, and read his recent work published in Chemical Science below:
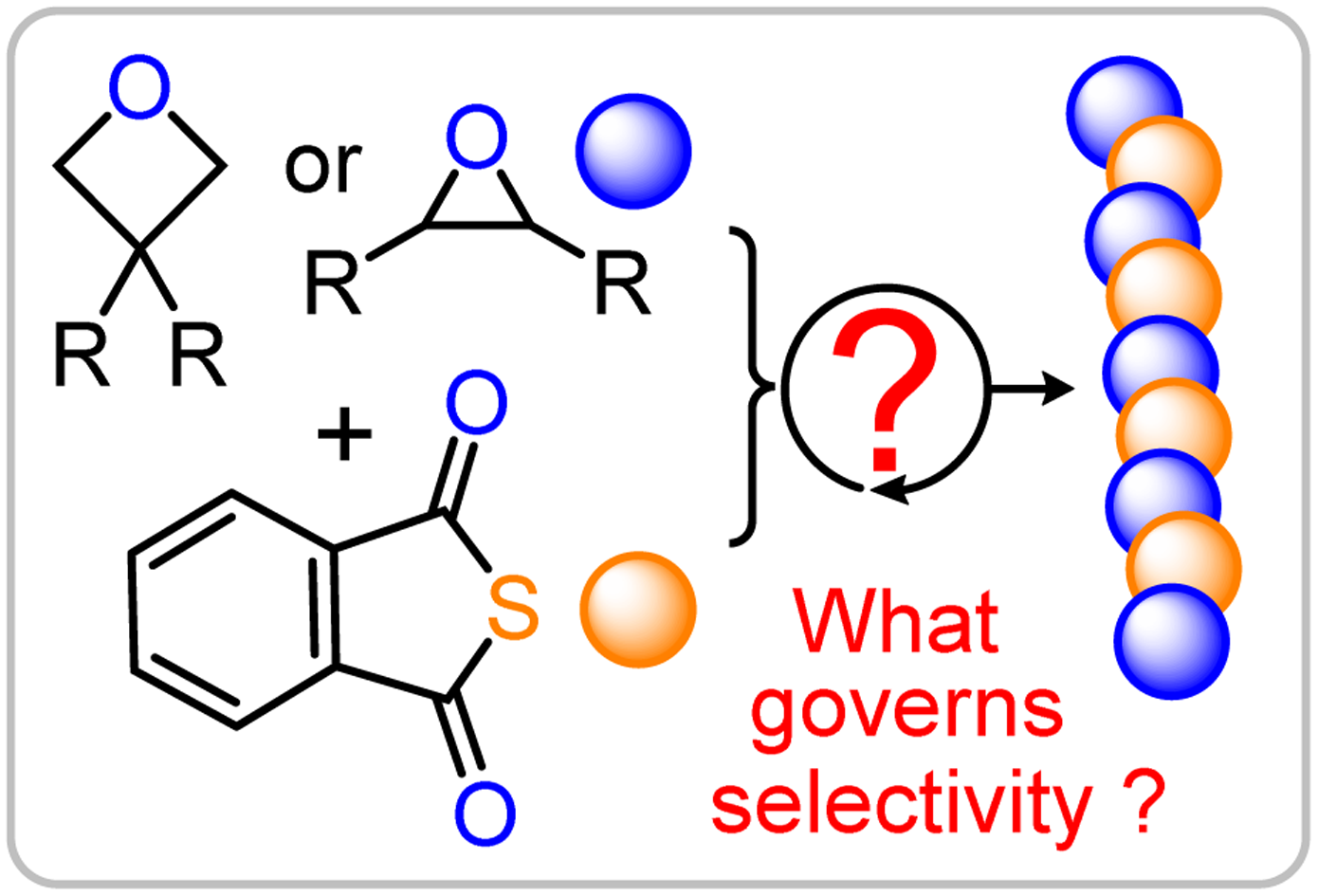
Entropy in catalyst dynamics under confinement Jun Cheng et al. Chem. Sci., 2024,15, 18303-18309
Spatial correlation of desorption events accelerates water exchange dynamics at Pt/water interfaces Xiandong Liu, Jun Cheng et al. Chem. Sci., 2025, 16, 2325-2334
Decoding the influence of monomer structures on the electrical double layer of alkaline fuel cells Jun Cheng et al. Chem. Sci., 2025, 16, 13741-13748
Please join us in welcoming Professors Yanai and Cheng to the journal!








")