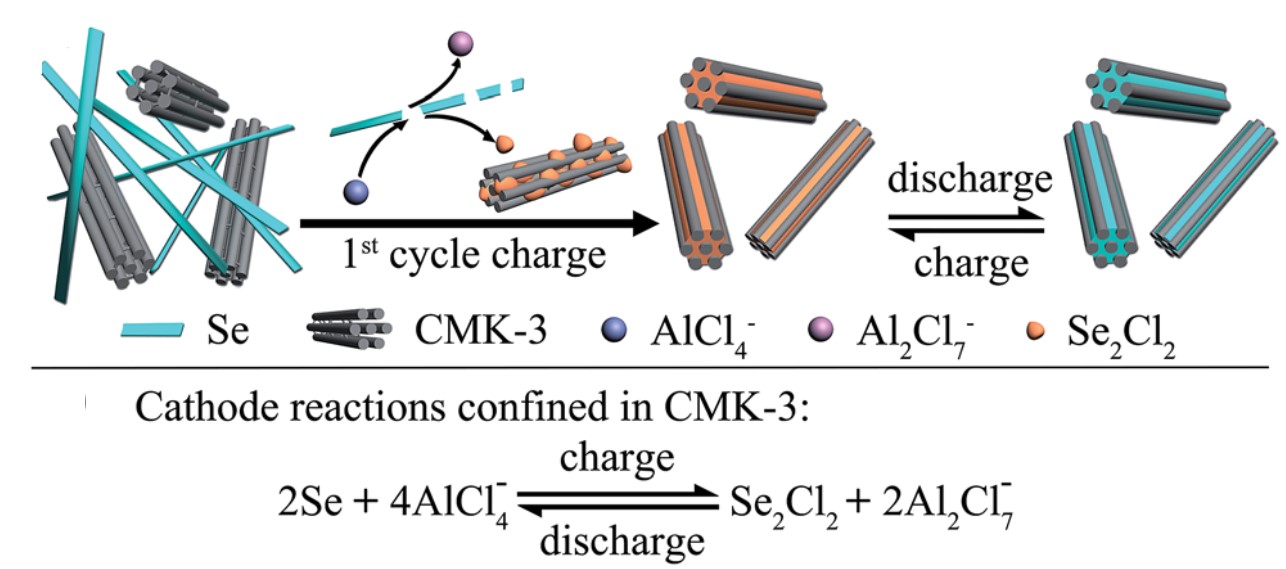
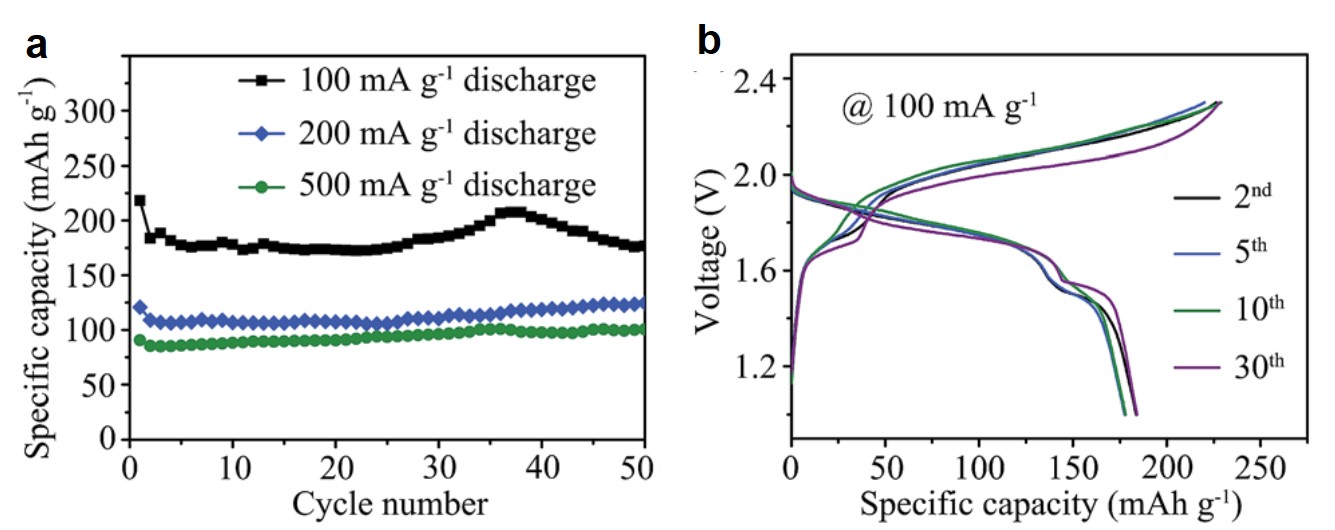
Dye-sensitized solar cells (DSSCs) are electrochemical devices that can convert solar energy into electricity. The critical component of a DSSC is its dye molecules which are covalently adsorbed on the electrodes of the DSSC. These molecules are responsible for light absorption and energy conversion in the device. DSSCs are more economical than commercial Si-based solar cells, but their lifetimes are limited (~6 years vs. 20-30 years of Si-based counterparts).
A research team from Xiamen University, China, recently demonstrated in Chemical Science (DOI: 10.1039/D0SC00588F) that the anchoring stability of the dyes determined the longevity of DSSCs. They specifically studied N719, a Ru-containing dye, adsorbed on three different crystal facets of rutile TiO2 (electrode). N719 adsorbed on the TiO2(111) facets was the most stable among all the facets studied.
The researchers adopted surface-enhanced Raman spectroscopy (Fig.1) in their research, the setup of which involved two laser beams. One 405-nm laser excited the dye molecules to initiate energy conversion, and another 638-nm laser collected the Raman scattering signals at the dye/TiO2 interface. The obtained Raman spectra showed the peaks associated with the vibrations of N719.
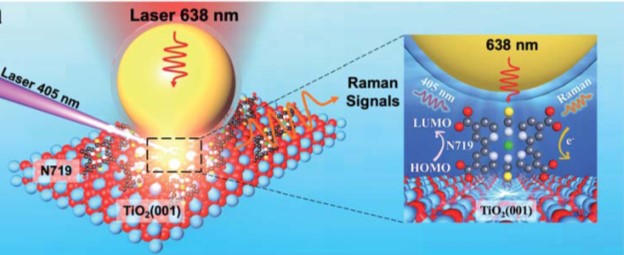
Figure 1. The experimental setup. The 405-nm laser excites N719 dye molecules adsorbed on rutile TiO2. The 638-nm laser probed the Raman scattering signals of N719. The Au nanoparticle (yellow sphere) enhances the Raman signal intensity.
The Raman spectra revealed that the adsorption stability of N719 depended on the crystal facet of TiO2. For TiO2(001), after illumination for 36 min, the Raman peaks of N719 gradually diminished (Fig. 2a), indicating that the dye molecules were either detached from TiO2 or decomposed. A similar trend was observed for N719 on TiO2(110) (Fig. 2b). Mass spectroscopy detected that the electrolytes after 36-min illumination contained N719 molecules missing an S atom. This result indicated that the C=S bond of N719 was broken, leading to the loss of the dye. In contrast, N719 on TiO2(111) exhibited stable Raman signals during the identical illumination duration (Fig. 2c).
The different stability was ascribed to variation in the dissociation energy of the C=S bond. Density functional theory (DFT) simulation proved that the cleavage of the C=S bond on TiO2(111) had an energy barrier of 3.5 eV, about 1.0 eV and 1.5 eV higher than those on TiO2(110) and TiO2(001), respectively. The higher energy barrier suppresses bond dissociation and stabilizes the adsorption of N719.
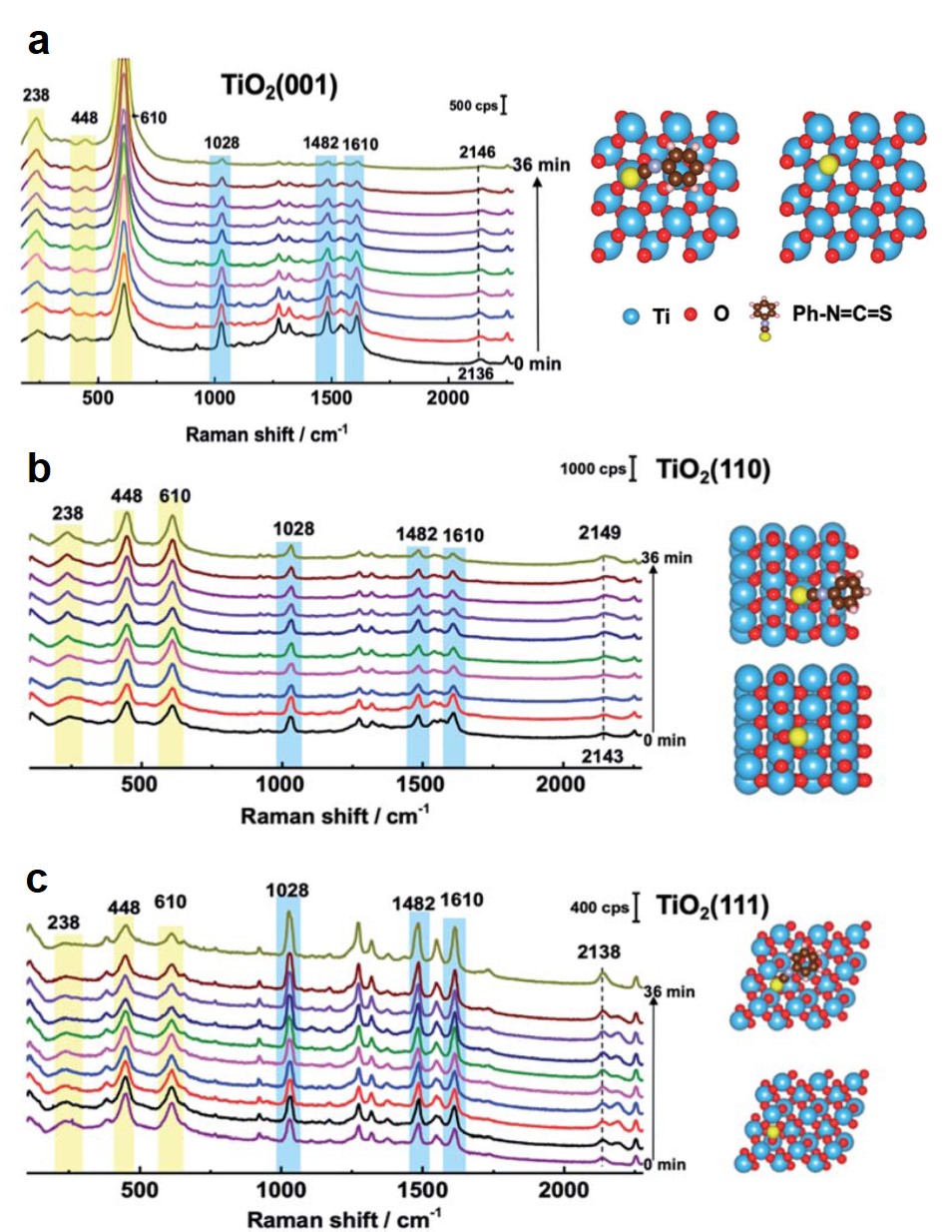
Figure 2. Raman spectra of N719 adsorbed on (a) TiO2(001), (b) TiO2(110), and (c) TiO2(111). Spectra were collected with an interval of 4 min. Peaks highlighted in yellow and blue are from TiO2 and N719, respectively. The schemes on the right show the simulated structures of dye-adsorbed (top) and desorbed (bottom) TiO2 facets. Ph–N=C=S represents N719 in simulation. The yellow spheres are S.
This work highlights the importance of dye positioning in promoting long-lasting performance of DSSCs.
For expanded understanding, please read:
Sheng-Pei Zhang, Jia-Sheng Lin, Rong-Kun Lin, Petar M. Radjenovic, Wei-Min Yang, Juan Xu, Jin-Chao Dong, Zhi-Lin Yang, Wei Hang, Zhong-Qun Tian, and Jian-Feng Li
Chem. Sci., 2020, DOI: 10.1039/D0SC00588F
Tianyu Liu acknowledges Zacary Croft at Virginia Tech, U.S., for his careful proofreading of this post.
About the blogger:
 Tianyu Liu obtained his Ph.D. (2017) in Chemistry from the University of California, Santa Cruz, in the United States. He is passionate about the communication of scientific endeavors to both the general public and other scientists with diverse research expertise to introduce cutting-edge research to broad audiences. He is a blog writer for Chem. Comm. and Chem. Sci. More information about him can be found at http://liutianyuresearch.weebly.com/.
Tianyu Liu obtained his Ph.D. (2017) in Chemistry from the University of California, Santa Cruz, in the United States. He is passionate about the communication of scientific endeavors to both the general public and other scientists with diverse research expertise to introduce cutting-edge research to broad audiences. He is a blog writer for Chem. Comm. and Chem. Sci. More information about him can be found at http://liutianyuresearch.weebly.com/.








")



 Tianyu Liu obtained his Ph.D. (2017) in Physical Chemistry from University of California, Santa Cruz in the United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at
Tianyu Liu obtained his Ph.D. (2017) in Physical Chemistry from University of California, Santa Cruz in the United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at