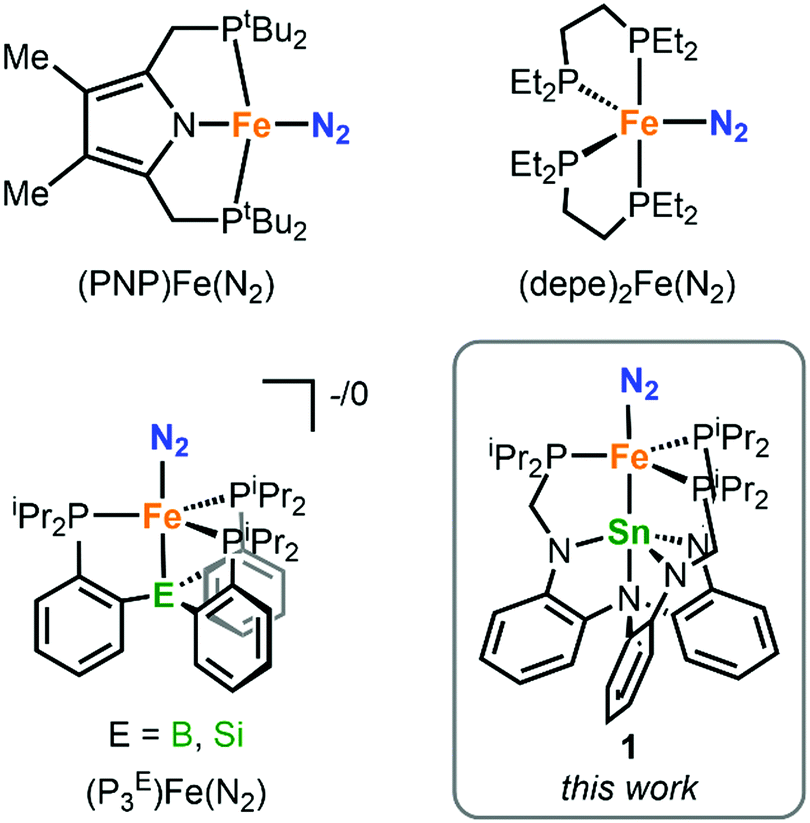
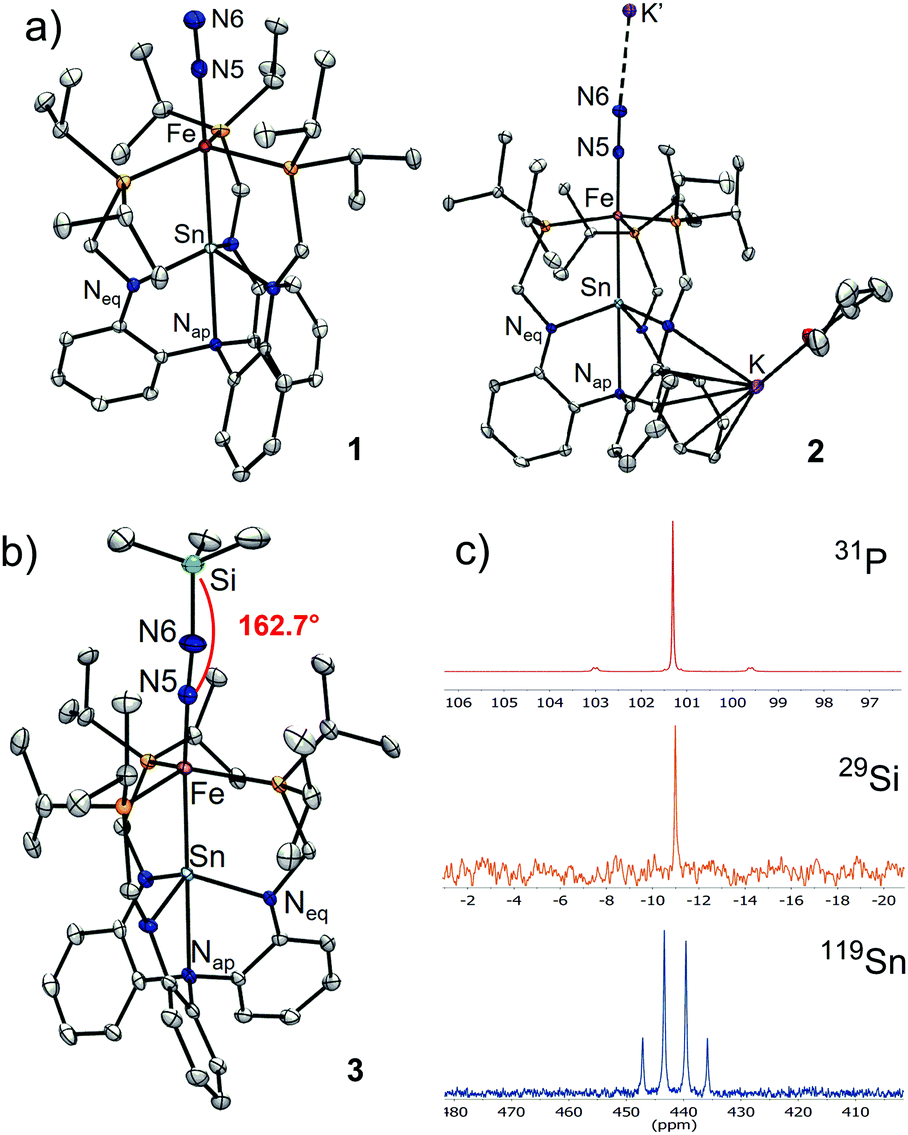
This week, we are bringing you more from #ChemCommMilestones – we spoke to Alex Murray about becoming a first-time indepedent author and publishing with our journal. Read Alex’s #ChemComm1st article: Ionicity-dependent proton-coupled electron transfer of supramolecular self-assembled electroactive heterocycles.
Find out more about Alex in our interview with him below.
What are the main areas of research in your lab and what motivated you to take this direction?
It’s probably easier to start with the second part: I trained as very much an organic chemist, but then moved to the US and learned electrochemistry as a postdoc, but I really am interested in redox-active small molecules more than anything else. The main applications that spring from this that we are researching are firstly new organic redox-flow batteries, and secondly the use of these small organic molecules as homogeneous electrocatalysts, especially with interesting interfacial behaviour.
Can you set this article in a wider context?
Well, this started as a side project really. I was watching a talk by a student from the Hiscock group in my first few weeks as a PI, and I was quite fascinated by their self-associating quinones. There has been so much fantastic and complex work done on the nuances of the electrochemistry of even quite simple quinones so I was really intrigued how these ‘quinone-SSAs’ would behave. What we found, broadly, is that the larger the size of the self-associated species, the more it behaved like a quinone in unbuffered or organic solution, so there appears to be some sort of barrier to proton transfer. But this is interesting, because for this system self-association makes the electron transfer faster, whereas often people have observed the opposite effect. I think with all the excitement about anthraquinones in organic redox flow batteries, the more unusual behaviours we know to look out for the better… and we are working on making other self-associated redox active heterocycles of course.
What do you hope your lab can achieve in the coming year?
Firstly we are following up on this collaboration, where we are interested in more complex supramolecular systems where we can control the self-association more readily. Secondly, we are hoping to make progress on both a new organic redox flow battery, and a new catalytic system we have in the works. It’s been a really tough few months, especially for my international PhD student who struggled to leave and return to her family, then struggled even harder to return to the UK. But things are looking up, and we are hoping to have more really exciting science to show within the next year.
Describe your journey to becoming independent researcher.
Since about a year into my PhD I think this is always something I’d wanted to do, though I was aware it’s not an easy road to say the least! I think the turning point for me was learning about electrochemistry – I really felt the confidence of having a more unique skill set than when I’d been trying to write ‘pure organic’ chemistry proposals, so my personal advice to PhD students and postdocs who want to be independent researchers is definitely to try and learn something very different – find a new field and learn to talk to them, but in a different way than how they talk to one other.
What is the best piece of advice you have ever been given?
“Why not, and what’s the worst that could happen”… this is good advice for crazy scientific ideas (the famous ‘Friday afternoon reaction’), but not in all aspects of life…
Why did you choose to publish in ChemComm?
We chose ChemComm because of the fast publication time, good support for early career researchers and positive previous experiences with the publication process at ChemComm and the RSC in general. Also this paper really sits at the interface of (organic) electrochemistry and supramolecular chemistry so it definitely made sense to go for a journal with a pretty general readership.
 |
Alex was born in Hull, UK in 1989, and obtained an MChem in Chemistry from the University of Sheffield in 2011, with a year of this degree undertaken at Monash University (Australia). He then carried out research in redox organocatalysis at the University of Bath, working in the group of Dr. Dave Carbery, receiving a PhD in 2015. This also included a CASE placement at GlaxoSmithKline. Alex then moved to the University of Nottingham, working for one year in the group of Professor Chris Moody on generating sp3-rich scaffolds for medicinal chemistry. Alex then moved to the US, receiving a Dreyfus Postdoctoral Fellowship to work on electrochemical catalysis in the group of Prof. Yogesh Surendranath at MIT. In May 2018 Alex returned to the UK and was appointed as a Lecturer at the University of Kent. |
Don’t miss more #ChemComm1st articles in our collection ChemComm Milestones – First Independent Articles.








")