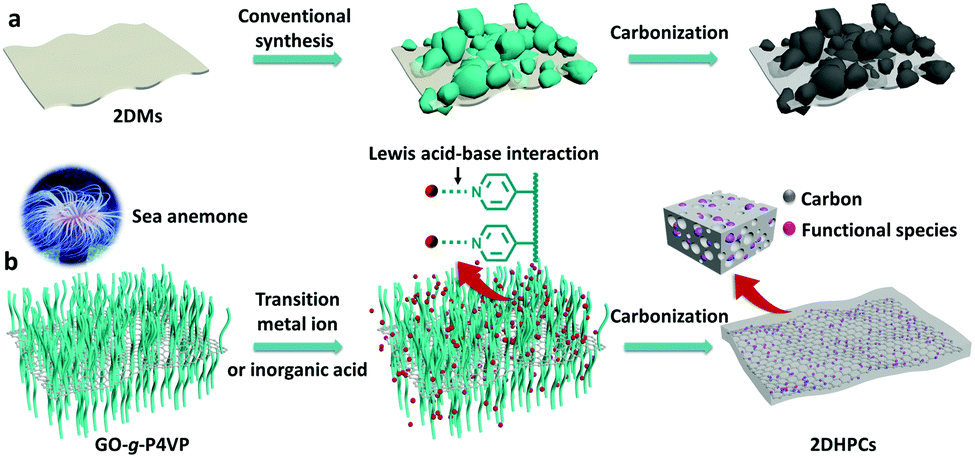
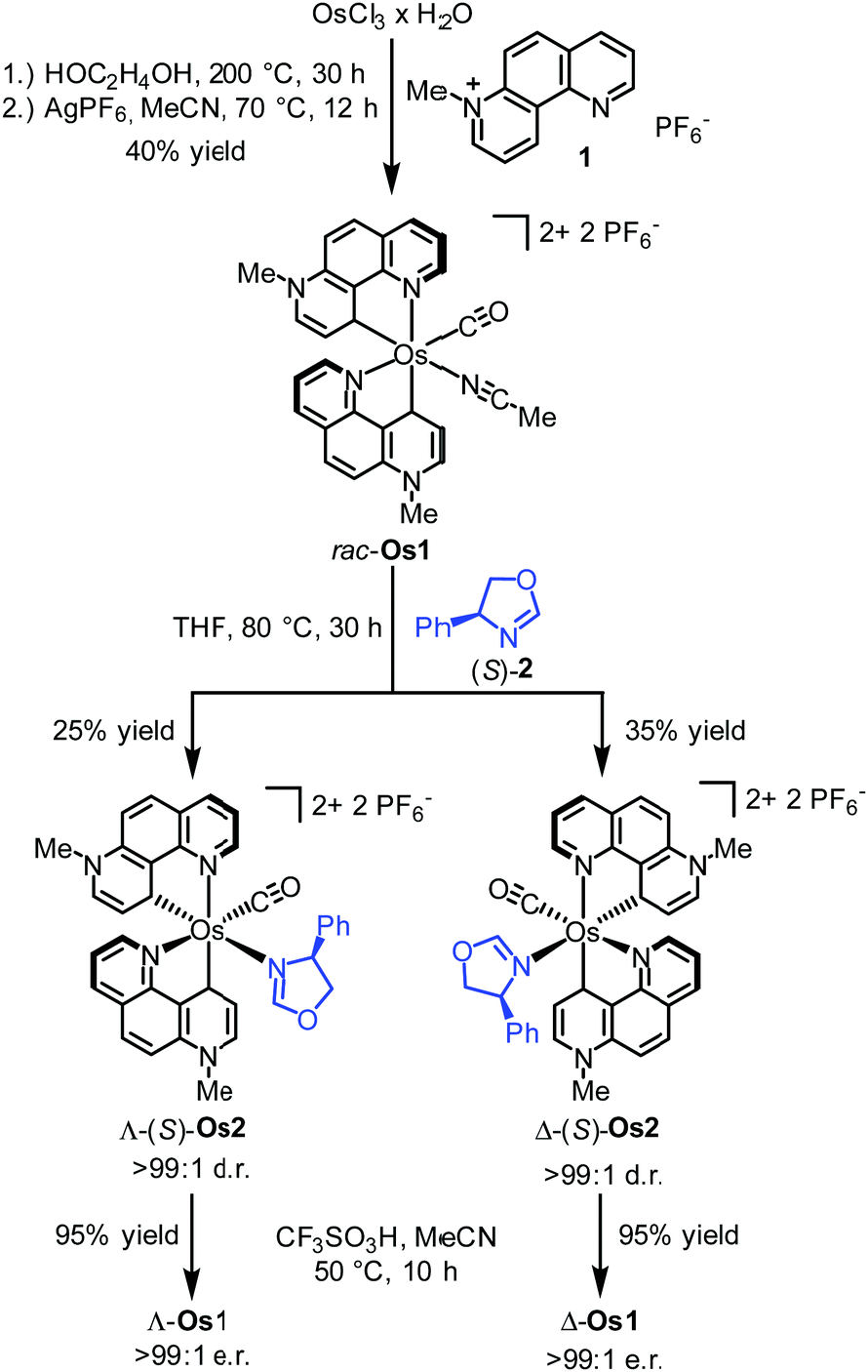
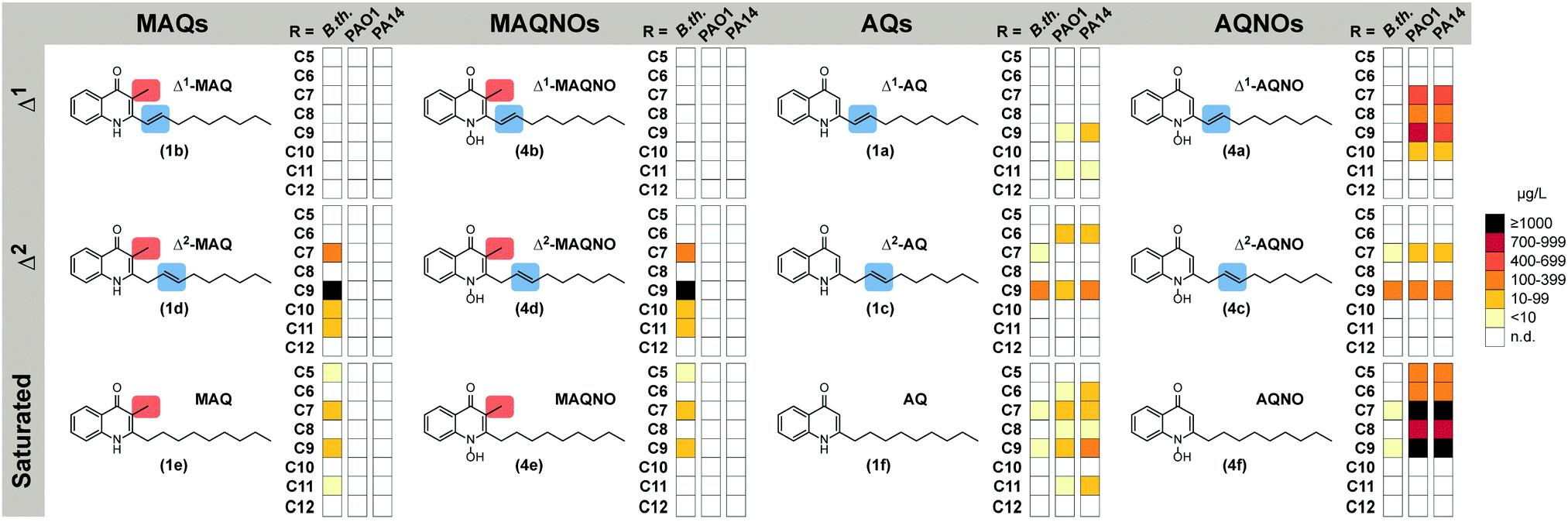
How can you study an unstable biomolecule? One research team’s answer to this is to surround it in a protective cradle. The molecules in question here are cysteine sulfenic acids (Cys-SOH); although they are crucial in various cellular processes and biological functions, they have proven elusive for isolated small-molecule studies due to their instability. Researchers in Japan have now reported the first synthesis and isolation of a stable small molecule cysteine sulfenic acid by employing a protective cradle for the reactive Cys-SOH unit. This bio-inspired design mimics the supramolecular protein environments that can stabilise Cys-SOH residues in situ.
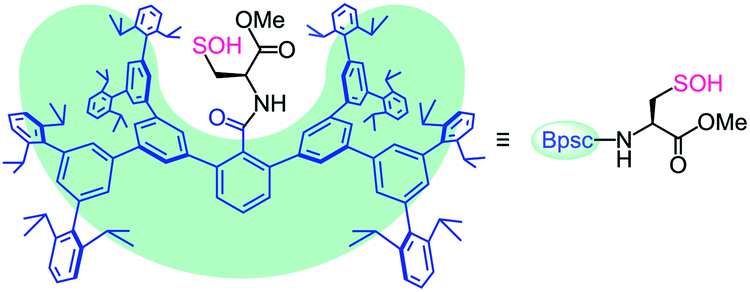
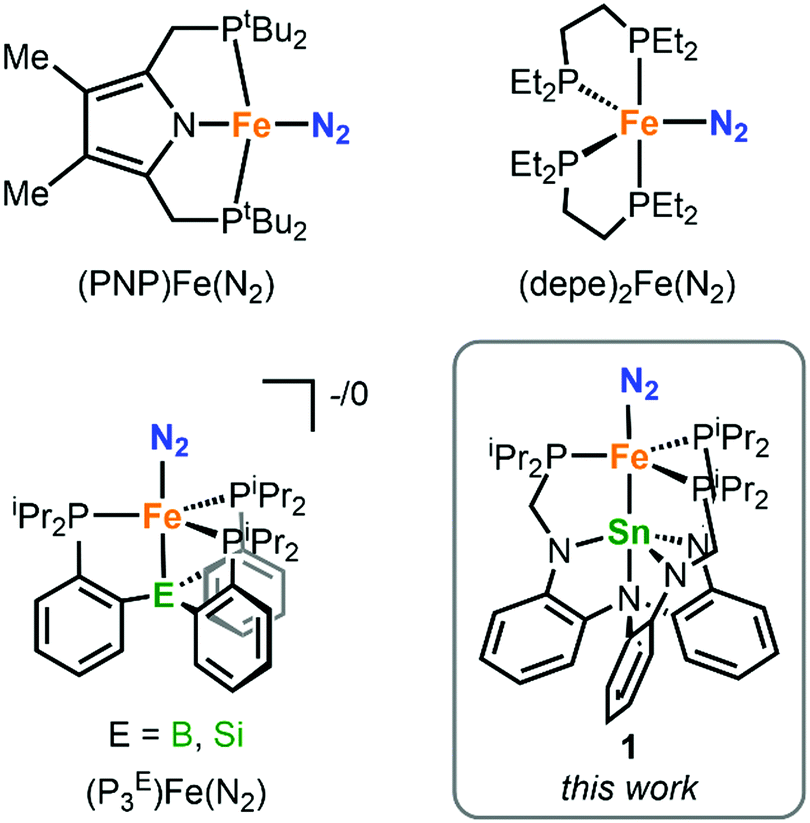
Figure 1. The chemical structure of the protective cradle for cysteine-derived reactive intermediates, including Cys-SOH (framework in blue, cysteine in black).
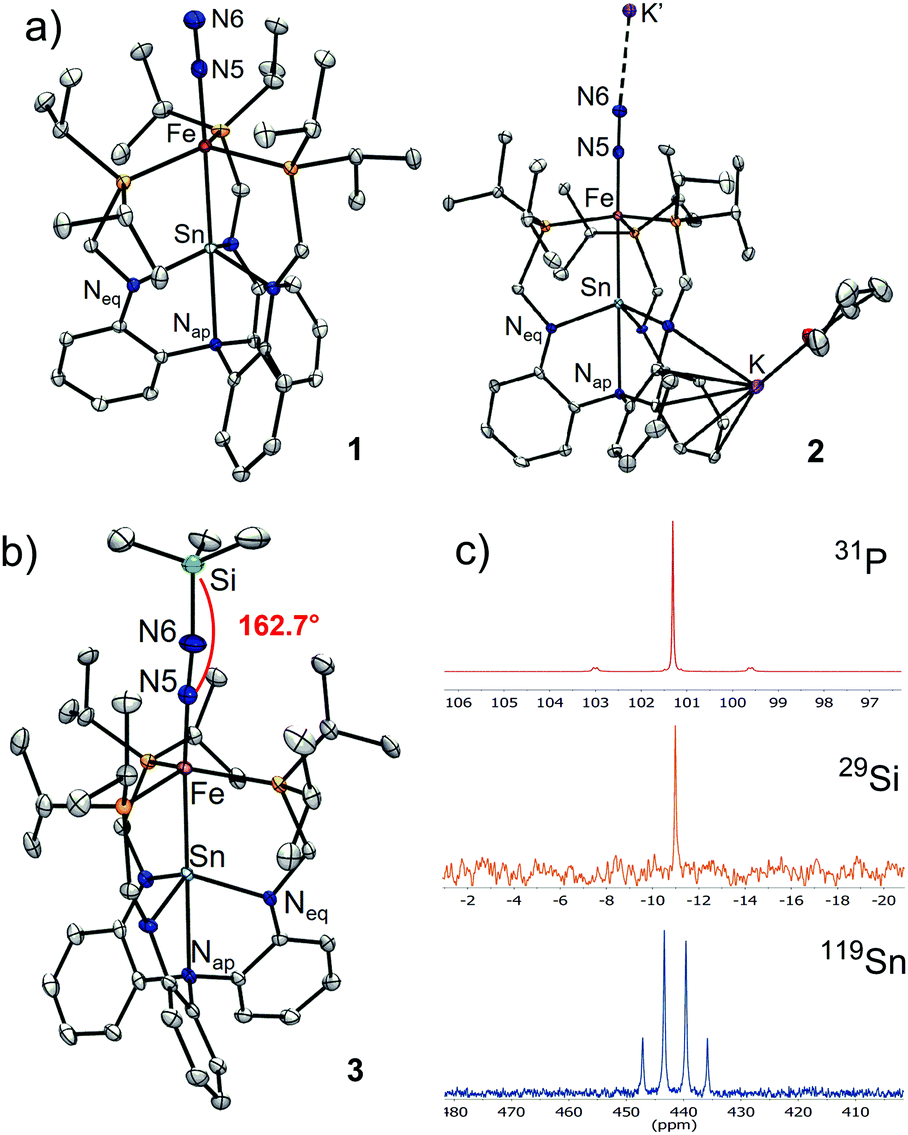
The researchers utilised the m-phenylene dendrimer framework from their previous studies as the molecular cradle to encapsulate cysteine intermediates for further study (Figure 1). A cysteine unit was first installed into the framework (denoted as Bpsc) via the central benzoyl group to generate the cysteine thiol residue, Cys-SH. The cradled Cys-SH was then converted to the cysteine sulfenic acid by treatment with H2O2, to give the isolable small molecule Cys-SOH (compound 6). The cysteine sulfenic acid 6 proved to be air and thermally stable, and the researchers characterised 6 using NMR and IR spectroscopies, as well as X-ray crystallographic analysis that definitively revealed its structure (Figure 2).

Figure 2. The crystal structure of the isolated Cys-SOH small molecule 6, cradled by the Bpsc framework (in green).
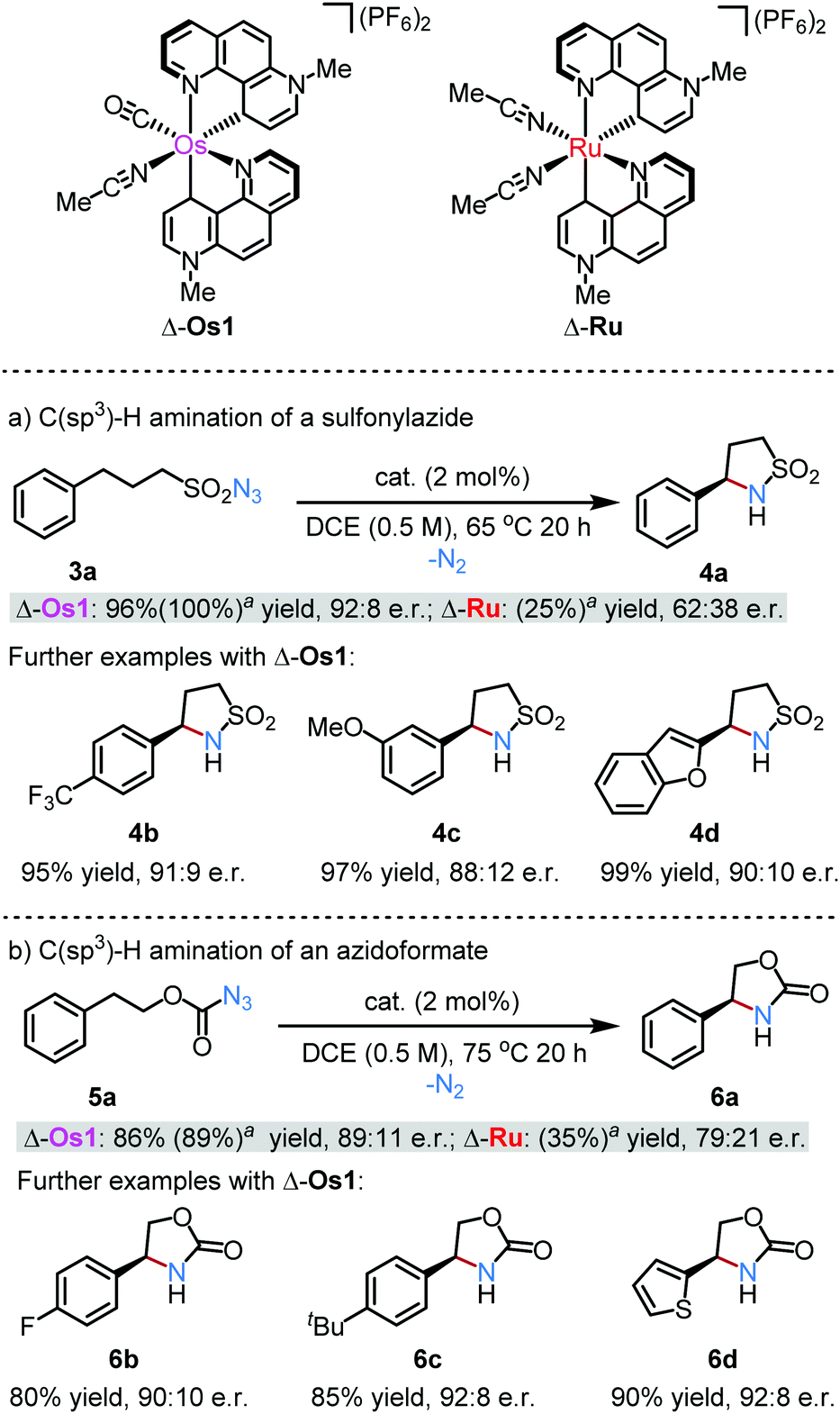
Once the structure of the cradled cysteine sulfenic acid 6 was established, the researchers investigated its biologically relevant reactivity. The reaction of Cys-SOH with a thiol is important for redox regulation and protein folding, and the researchers observed that 6 did indeed react with the thiol N-acetylcysteine methyl ester to yield the expected disulfide product. Although this reaction initially proved slow, the researchers were able to show that this was not due to steric hinderance from the Bpsc cradle. Optimisation of this reaction revealed that the addition of triethylamine significantly enhanced the reactivity, and this suggests that the presence of basic residues in the vicinity of cysteine sulfenic acids are important for biological disulfide formations.
In addition to the disulfide formation, the researchers also investigated the reactivity of their cysteine sulfenic acid 6 towards reduction and trapping experiments. The Cys-SOH 6 could be reduced by DTT (dithiothreitol) to regenerate the Cys-SH thiol residue, demonstrating the reversibility of the redox processes between Cys-SH and Cys-SOH within the cradle. The researchers also looked at trapping the Cys-SOH residue using various chemical probes to mimic trapping experiments used for protein-generated cysteine sulfenic acids. They observed the expected formation of the thiol adducts, and again noticed enhanced reactivity in the presence of triethylamine.
Overall, the researchers have demonstrated the first example of an isolable and stable cysteine sulfenic acid that is enabled by a cradling framework. The protective cradle was shown to not impact the biologically-relevant reactivity of the Cys-SOH residue, and ultimately, this framework could serve as a useful biorepresentative model for future studies on Cys-SOH to further understand its behaviour.
To find out more, please read:
Isolable small-molecule cysteine sulfenic acid
Tsukasa Sano, Ryosuke Masuda, Shohei Sase and Kei Goto*
Chem. Commun., 2021, 57, 2479-2482
About the blogger:
 Dr. Samantha Apps recently finished her post as a Postdoctoral Research Associate in the Lu Lab at the University of Minnesota, USA, and obtained her PhD in 2019 from Imperial College London, UK. She has spent the last few years, both in her PhD and postdoc, researching synthetic nitrogen fixation and transition metal complexes that can activate and functionalise dinitrogen. Outside of the lab, you’ll likely find her baking at home, where her years of synthetic lab training has sparked a passion in kitchen chemistry too.
Dr. Samantha Apps recently finished her post as a Postdoctoral Research Associate in the Lu Lab at the University of Minnesota, USA, and obtained her PhD in 2019 from Imperial College London, UK. She has spent the last few years, both in her PhD and postdoc, researching synthetic nitrogen fixation and transition metal complexes that can activate and functionalise dinitrogen. Outside of the lab, you’ll likely find her baking at home, where her years of synthetic lab training has sparked a passion in kitchen chemistry too.








")