Crystalline MOF structures with high porosity serve well for a range of applications including shape selective catalysis, gas storage or capture, and drug delivery. These entail the prominence of new methodologies for the synthesis of MOFs, either with unusual metal ion oxidation states or already existing molecular counterparts. In this vein, researchers from TU Denmark have used a ligand transferability strategy for the reaction of homoleptic carbonyl complexes of the Group 6 elements, M(CO)6 (M = Cr, Mo, W), with neutral bridging ligands for the formation of heteroleptic M(CO)6-nLn complexes. For the first time, they have presented the synthesis and characterization of a series of MOFs obtained from M(CO)6 (M = Cr, Mo, W) through the substitution of the ditopic pyrazine (pyz) ligand.
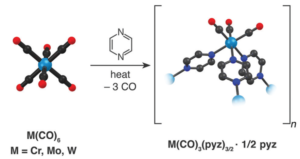 Scheme 1: Synthesis of Cr, Mo, and W.
Scheme 1: Synthesis of Cr, Mo, and W.
The synthesis follows reaction at an elevated temperature between M(CO)6 (M = Cr, Mo, W) and an excess of the pyz ligand in a sealed ampoule (Scheme1), leading to dark coloured crystalline products. The reaction is highly favourable for the Mo complex, due to the low dissociation energy of the first Mo–CO bond (119 kJ mol-1), and a reaction temperature of 150°C produced a high yield of crystals. The same reaction conditions provided a minute amount of product for M = Cr and no indication of reactivity for M = W. However, a higher temperature of 200°C lead to the production of the W complex due to the high dissociation energy of the W-CO bond (142 kJ mol-1).
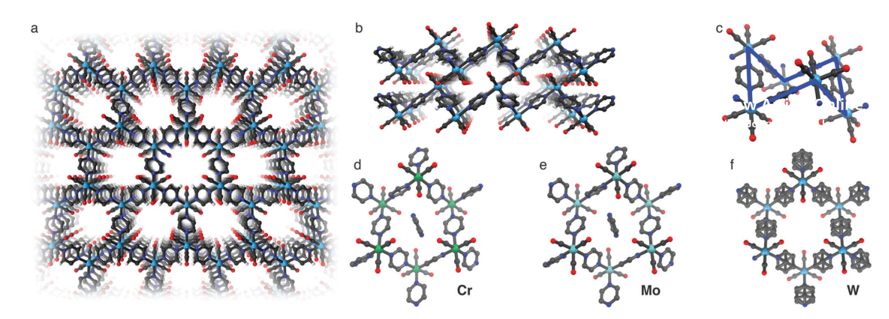
The single-crystal X-ray diffraction pattern of the dark shiny crystals suggests that the composition is fac-M(CO)3(pyz)3/2.1/2 pyz (with M = Cr, Mo, W) (Fig. 1). Cr and Mo crystallize in the triclinic P1 space group, whereas W crystallizes with higher symmetry in the monoclinic C2/m space group. The crystal structure predicts that the three remaining carbonyl ligands reach into the voids of the hexagonal tiling. The hexagon is in chair conformation leading to <M–M–M angles approaching 90° with hexagonal pore channels of roughly 5.5–6.5 Å. The hexagon for W is nearly equilateral in nature compared to Mo, which exhibits unequal hexagon edges.
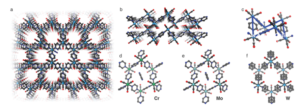 Fig 1: Single crystal structure of Cr, Mo, and W: (a) View of the hexagonal pore channels in W. (b) Stack of coordination layers in W. (c) Chair conformation of the hexagonal arrangement shown for W. The solvent molecules inside the pores and the disorder are not shown for clarity in (a–c). (d–f) Hexagonal fragments of Cr, Mo, and W together with the pyz molecule contained in the pores (not visible for W due to solvent mask, see methods). The positional disorder of the pyz molecules in W is also shown.
Fig 1: Single crystal structure of Cr, Mo, and W: (a) View of the hexagonal pore channels in W. (b) Stack of coordination layers in W. (c) Chair conformation of the hexagonal arrangement shown for W. The solvent molecules inside the pores and the disorder are not shown for clarity in (a–c). (d–f) Hexagonal fragments of Cr, Mo, and W together with the pyz molecule contained in the pores (not visible for W due to solvent mask, see methods). The positional disorder of the pyz molecules in W is also shown.
The typical IR absorption band related to M-CO stretching is explored for characterizing the complexes. A red shift of ~310 cm-1 in the spectra of the average carbonyl stretching band was obtained, which is approximated as a ~25% decrease of the force constant associated with the C–O bond. The thermogravimetric analysis showed that the Mo complex has the highest thermal stability with an onset of degradation around 180°C, whereas Cr and W underwent significant mass loss from around 150°C and 130°C, respectively. A similar trend is observed for the stability of the complexes in atmospheric air.
These first examples of structurally characterized zero-valent MOFs with metal nodes derived from metal carbonyls could be attractive architectures for the exploration of catalytic applications as they facilitate the possibility for the non-destructive removal of pore-filling molecules.
For further details, please go to:
Zero-valent metals in metal–organic frameworks: fac-M(CO)3(pyrazine)3/2
Laura Voigt, Rene´ Wugt Larsen, Mariusz Kubus and Kasper S. Pedersen*
Chem. Commun., 2021, 57, 3861
About the writer:
Damayanti Bagchi, PhD, is a postdoctoral researcher in Irene Chen’s lab at University of California, Los Angeles, United States. She has obtained her PhD in Physical Chemistry from Satyendra Nath Bose National Centre for Basic Sciences, India. Her research is focused on spectroscopic studies of nano-bio interface and phage therapy. She is interested in science communication and science policy-diplomacy. She enjoys travelling and experimenting with various cuisines!
You can find her on Twitter @DamayantiBagchi









")