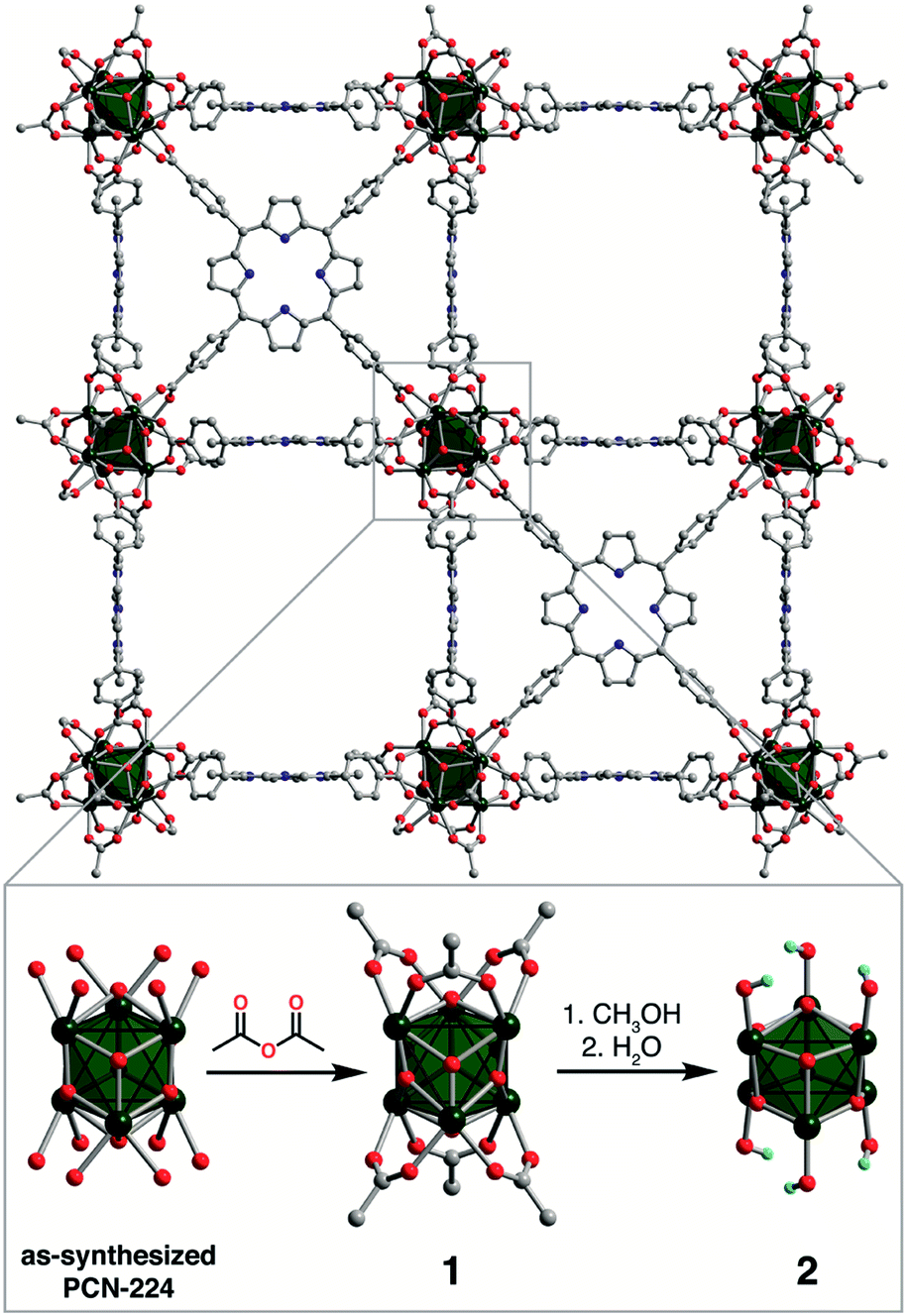
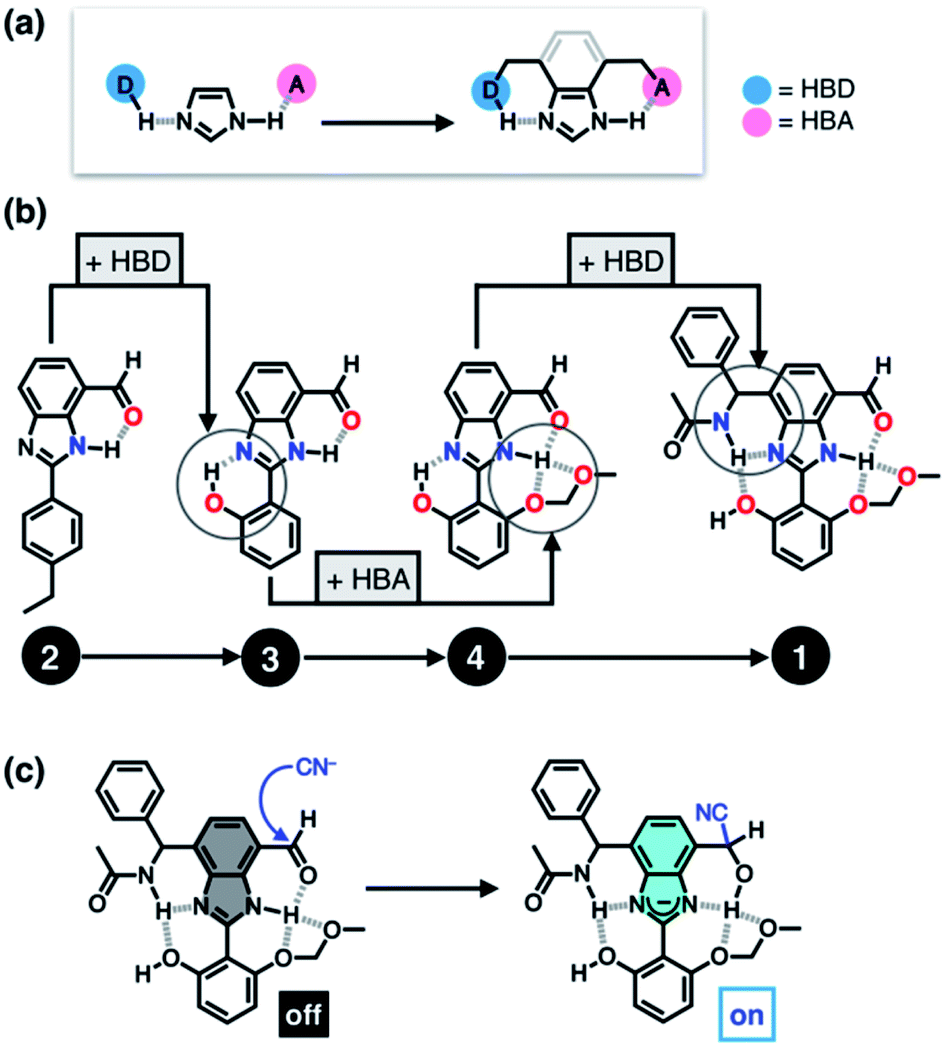
The mighty gear is essential in machines. Even when scaling down the size of the machine, like from cars to small wristwatches, gears are necessary to transmit motion and mechanical power across the system. Machines can be decreased in size all the way to the nanoscale with molecular machines, where individual molecules can produce mechanical motion in response to external stimuli. Just as in macroscopic machines (e.g. cars), the addition of gears to nanomachines is needed for creating more complex assemblies with controlled motion, extending the applications of these molecules beyond the fundamental.

Figure 1. A schematic representation of the design for trains of molecular gears.
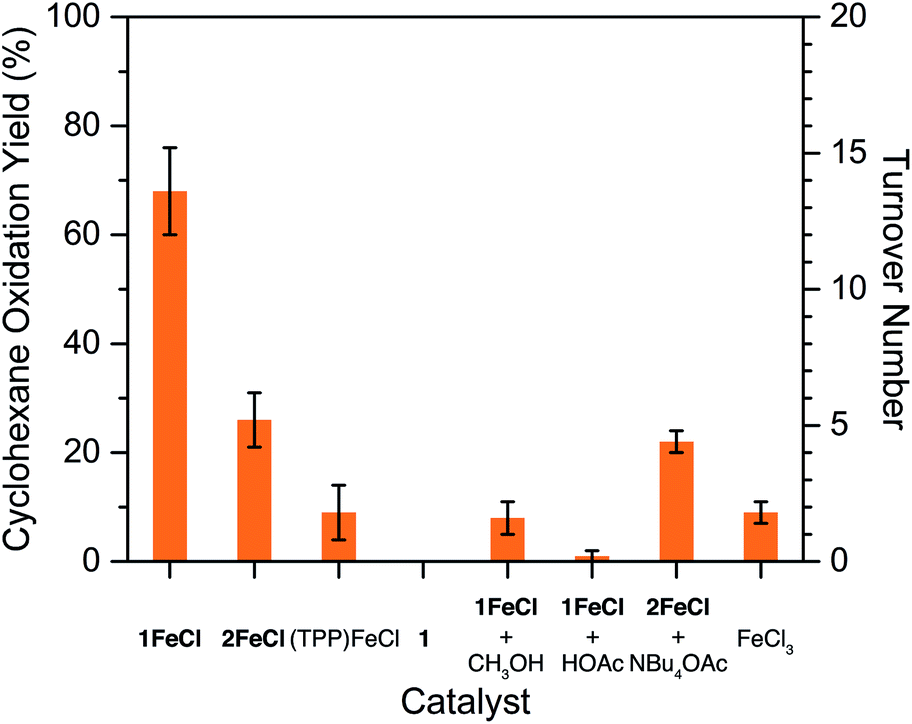
A team of researchers from France and Japan have now reported a series of molecular gears, with the aim of achieving correlated motion within trains of gears across a surface (Figure 1). To achieve this correlated motion, the researchers designed desymmetrised organometallic molecular gears based around star-shaped ruthenium piano-stool complexes. These molecular gears incorporated a facially capping hydrotris(indazolyl)borate ligand at one end, which both anchors the complex to the surface and lifts the central metal centre away to enable a rotational axis around the ruthenium. At the other end, the molecular gears have a cyclopentadienyl core to act as the cogwheel, functionalised with bulky groups that mimic the teeth that allow correlated motion between the gears (Figure 2). The researchers set out to make these molecular gears with lower symmetry to allow for detailed on-surface mechanical studies, by changing one of the five teeth (i.e. the functionalised groups around the cyclopentadienyl core) to include a steric or chemical tag– this is shown in Figures 1 and 2 by the green section.
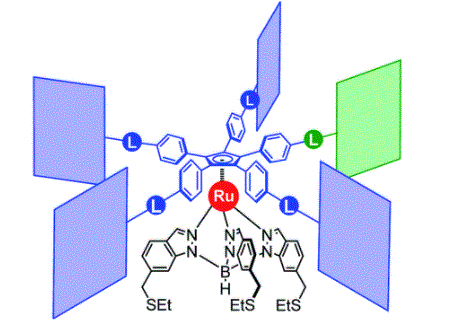
Figure 2. Chemical structure of the molecular gears, with the anchoring ligand in black beneath the ruthenium centre and the rotating cogwheel cyclopentadienyl ligand in blue. The rectangles represent the teeth of the cogwheel as the bulky groups added to the central cyclopentadienyl core, where one of the five teeth (coloured green rather than blue) is sterically or chemically changed to lower the symmetry.
The researchers developed a modular synthetic approach to achieve desymmetrisation of the star-shaped ruthenium molecular gears, based on post-functionalisation of the central cyclopentadienyl core with Ni(II) porphyrins to act as the teeth of the cogwheels. They used an unsymmetrical 1,2,3,4,5-penta(p-halogenophenyl)cyclopentadienyl as the core; the p-halogenophenyl groups are all pre-activated to allow for further functionalisation, but one of the five is a p-iodophenyl group that chemoselectively reacts over the other four p-bromophenyl groups. Scheme 1 shows a sequential synthetic route towards one of the desymmetrised molecular gears: the p-iodophenyl group is first functionalised with a unique porphyrin (shown in green), before subsequent functionalisation of the four other p-bromophenyl groups with the same porphyrins (shown in blue), all using palladium-catalysed cross-coupling reactions.
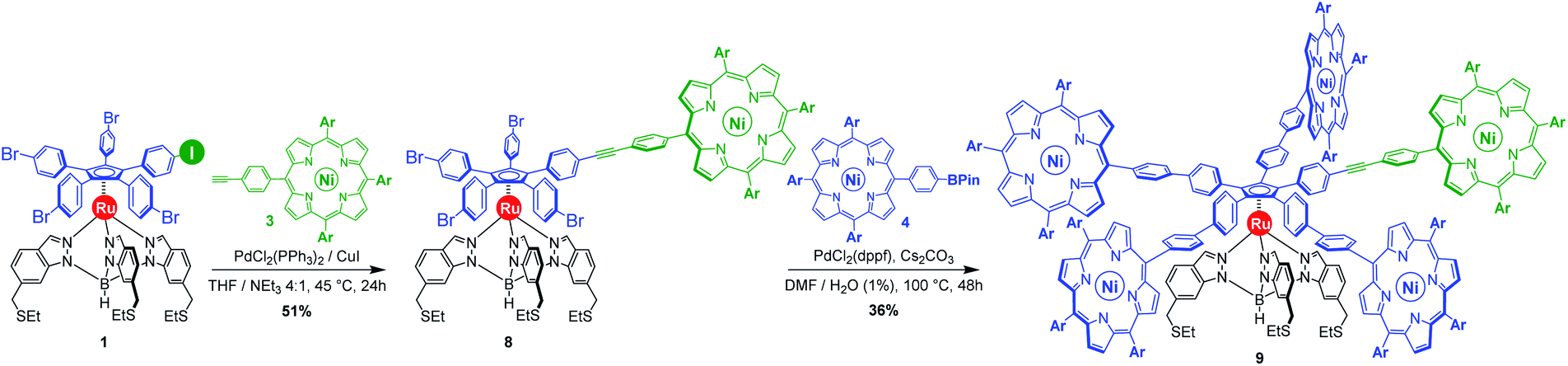
Scheme 1. An example synthetic route towards desymmetrised molecular This example shows a sterically tagged cogwheel, where the unique porphyrinic tooth (in green) contains a longer linker than the four other teeth (in blue).
The researchers varied their approach to changing the unique porphyrinic tooth for the molecular gear, using either steric tagging (with one longer linker between the porphyrin and p-halogenophenyl group) or chemical tagging, using either one distinct electron-deficient porphyrin (achieved by using p-cyanophenyl substituents on the tetrapyrrole core) or one distinct metal porphyrin (Zn(II) instead of Ni(II)). The synthesised desymmetrised molecular gears were characterised using spectroscopic and electrochemical techniques, and the researchers are currently undertaking further mechanical studies to understand the correlated motion of these gears on surfaces.
To find out more, please read:
Desymmetrised pentaporphyrinic gears mounted on metallo-organic anchors
Seifallah Abid, Yohan Gisbert, Mitsuru Kojima, Nathalie Saffon-Merceron, Jérôme Cuny, Claire Kammerer* and Gwénaël Rapenne*
Chem. Sci., 2021, Advance Article
About the blogger:
 Dr. Samantha Apps recently finished her post as a Postdoctoral Research Associate in the Lu Lab at the University of Minnesota, USA, and obtained her PhD in 2019 from Imperial College London, UK. She has spent the last few years, both in her PhD and postdoc, researching synthetic nitrogen fixation and transition metal complexes that can activate and functionalise dinitrogen. Outside of the lab, you’ll likely find her baking at home, where her years of synthetic lab training has sparked a passion in kitchen chemistry too.
Dr. Samantha Apps recently finished her post as a Postdoctoral Research Associate in the Lu Lab at the University of Minnesota, USA, and obtained her PhD in 2019 from Imperial College London, UK. She has spent the last few years, both in her PhD and postdoc, researching synthetic nitrogen fixation and transition metal complexes that can activate and functionalise dinitrogen. Outside of the lab, you’ll likely find her baking at home, where her years of synthetic lab training has sparked a passion in kitchen chemistry too.








")


![Cyclic voltammograms of [Ni(PPh2NPh2)2]2+ in acetonitrile](https://blogs.rsc.org/sc/files/2020/11/CS-Fig-1.gif)
![Redox cycle scheme for [Ni(PPh2NPh2)2]2+](https://blogs.rsc.org/sc/files/2020/11/CS-Fig-2.gif)