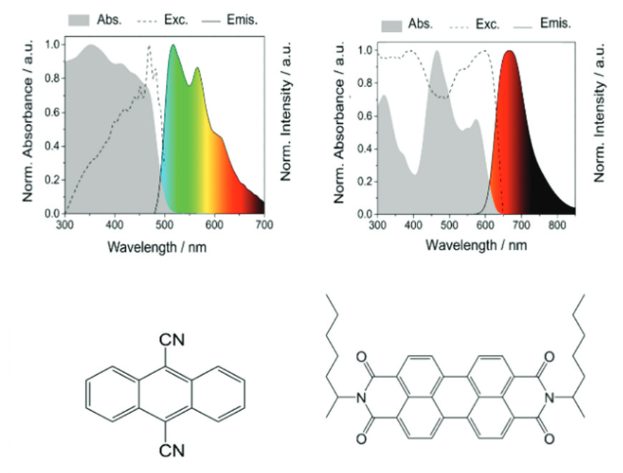
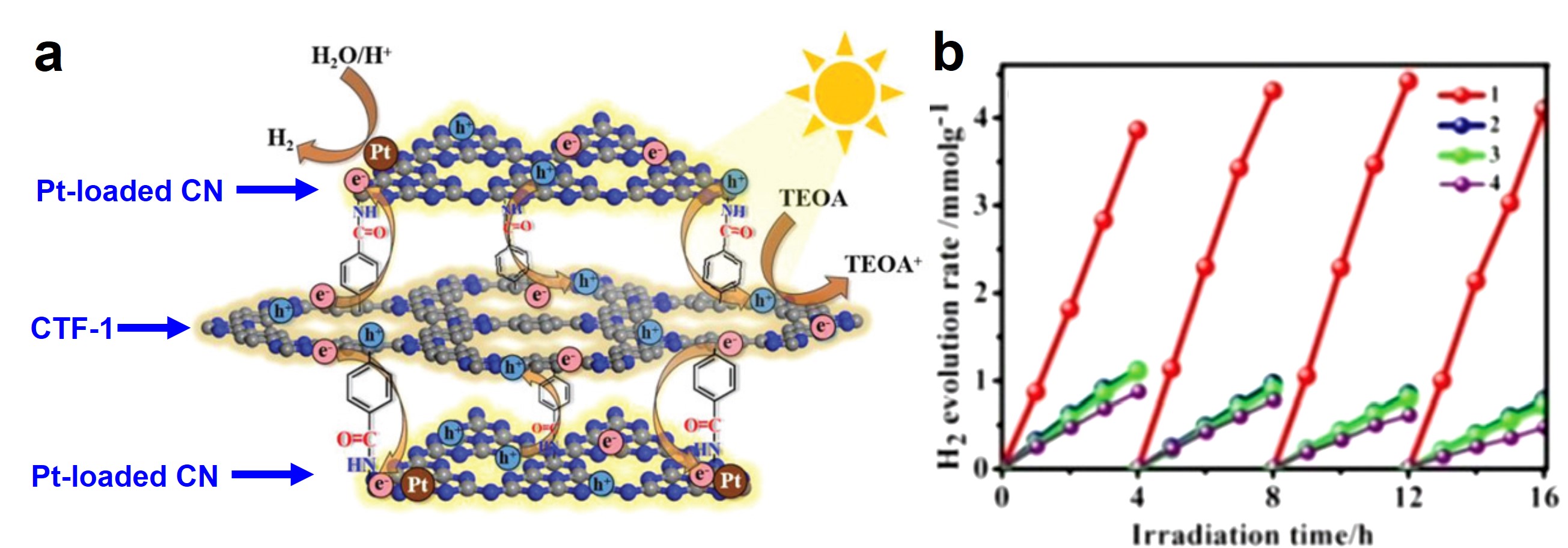
Lithium-metal batteries are a family of rechargeable batteries with higher charge-storage capacities than those of lithium-ion batteries. The boosted charge-storage performance of lithium-metal batteries is rooted in its anode material – Li metal, as it possesses an ultrahigh theoretical capacity (3860 mAh/g). However, the growth of dendrites on Li surfaces during charging could short-circuit batteries, cause combustion, and trigger explosions.
A research group led by Feng Li at the Institute of Metal Research, Chinese Academy of Sciences, recently devised a strategy to suppress the notorious Li dendrite growth in lithium-metal batteries. By tuning the composition of the electrolytes, the authors strengthened the coordination between Li+ and electrolyte solvents, which slowed the growth of Li dendrites. This work has been published in Chemical Communications (doi: 10.1039/C9CC07092C).
The researchers introduced an electrolyte additive, tetraethylene glycol dimethyl ether (TEGDME), as a coordination ligand to Li+. Compared to other components in the electrolyte, i.e., 1,2-dimethoxyethane (DME) and 1,3-dioxolane (DOL), TEGDME contains more oxygen atoms that can form multiple, robust coordination bonds with Li+. Specifically, density functional theory calculations showed that the binding energy between Li+ and electrolyte molecules increased by 0.31 eV after introducing TEGDME, reaching an absolute value of 4.93 eV. The enhanced binding force made the separation of Li+ from TEGDME (a prerequisite for Li-dendrite growth) energetically consuming and kinetically sluggish (Figure 1). These characteristics could decelerate Li-dendrite formation and elongate battery lifetimes.
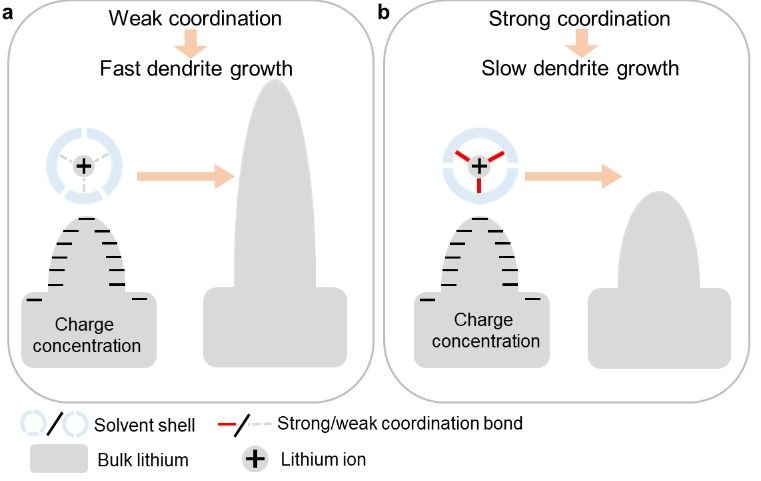
Figure 1. Lithium-dendrite growth in different electrolytes: (a) weak coordination with Li+ promotes fast dendrite growth while (b) strong coordination with Li+ decelerates dendrite formation.
To confirm the above idea, the authors assembled lithium batteries with TEGDME+DME+DOL or DME+DOL electrolytes. Cycling stability tests demonstrated that the battery with the TEGDME-added electrolyte survived 60 charge-discharge cycles at a current density of 1C, whereas the capacity of the battery without TEGDME rapidly decayed beyond 30 cycles under identical testing conditions (Figure 2a). Scanning electron microscopy images revealed that the number of rod-shaped Li dendrites on the anode in the TEGDME-added electrolyte (Figure 2c) was less than that in the TEGDME-free electrolyte (Figure 2b), further confirming that the enhanced cycling stability resulted from the Li-dendrite suppressing effect of TEGDME.
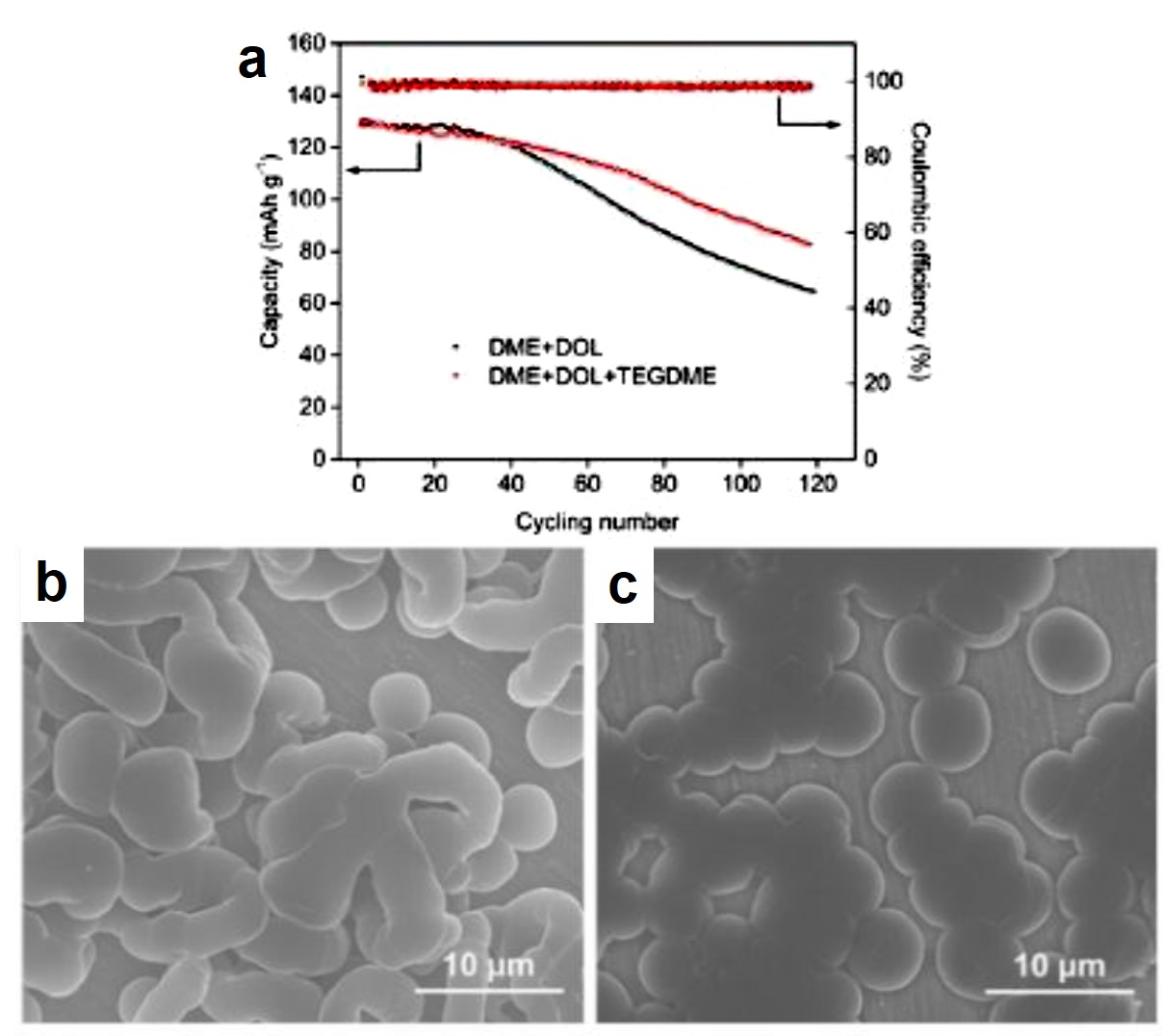
Figure 2. (a) Cycling stability performance of lithium-metal batteries with two different electrolytes. The cathode material in both batteries was lithium iron phosphate (LFP). (b and c) SEM images of the Li anode surface after charging in (b) DME+DOL and (c) DME+DOL+TEGDME electrolytes.
This work highlights the importance of tailoring the electrolyte composition for preserving the stability and safety of lithium-metal batteries.
To find out more, please read:
Suppressing Lithium Dendrite Formation by Slowing Its Desolvation Kinetics
Huicong Yang, Lichang Yin, Huifa Shi, Kuang He, Hui-Ming Cheng, and Feng Li
Chem. Commun., 2019, doi: 10.1039/C9CC07092C
Tianyu Liu acknowledges Xiaozhou Yang of Virginia Tech, the U.S., for his careful proofreading of this post.
About the blogger:
 Tianyu Liu obtained his Ph.D. (2017) in Chemistry from the University of California, Santa Cruz, in the United States. He is passionate about the communication of scientific endeavors to both the general public and other scientists with diverse research expertise as a way to introduce cutting-edge research to broad audiences. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at http://liutianyuresearch.weebly.com/.
Tianyu Liu obtained his Ph.D. (2017) in Chemistry from the University of California, Santa Cruz, in the United States. He is passionate about the communication of scientific endeavors to both the general public and other scientists with diverse research expertise as a way to introduce cutting-edge research to broad audiences. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at http://liutianyuresearch.weebly.com/.








")