Just in case you weren’t aware, it turns out that lithium-based batteries are kind of a big deal. While the Nobel-winning batteries have already revolutionized consumer electronics, further development requires batteries with even higher energy densities. Enter: lithium-oxygen batteries (LOBs) with theoretical energy densities of 3500 W h/kg. LOBs come in non-aqueous, aqueous, hybrid, and solid-state varieties based on their electrolytes. Given the previous safety issues for lithium-based batteries with liquid electrolytes (remember the exploding phones?), solid-state electrolytes have attracted substantial research attention. Specifically, Li1+xAlxGe2x(PO4)3, or LAGP, shows promise given its high Li+ transport number and electrochemical stability over a wide window. These solid-state electrolytes need to be combined with new catalytically active high surface area cathode materials that will not react with the lithium and degrade, a persistent issue with MOFs.
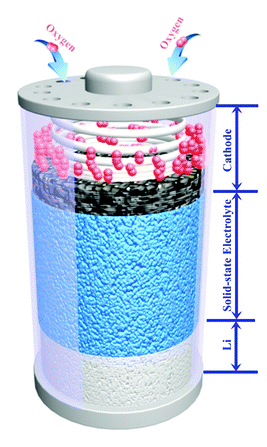
Figure 1. Schematic of an assembled all solid-state lithium-oxygen battery.
Researchers in China and Japan have combined LAGP electrolyte with NiCo2O4 (NCO) nanoflakes as the catalytically active cathode material. They then assembled full solid-state batteries, the structure of which is shown in Figure 1, for electrochemical and stability testing. The LAGP was prepared using previously established methods and found to exhibit the expected high stability and lithium mobility. To prepare the nanoflakes, the researchers annealed cobalt-based MOFs on a sacrificial carbon substrate then dipped them in a Ni(NO3)2 solution for nickel doping and annealed once more. This leaves the final nanostructured metal oxide, with the elemental composition confirmed by TEM elemental mapping. As a conveniently freestanding electrode material, the nanoflakes were then loaded in as the cathode.
Once assembled, the researchers tested the full all solid-state LOBs for stability and performance. They demonstrated high discharge capacity and electron transfer efficiency with charge and discharge potentials well within the electrochemical window of the LAGP electrolyte. These are attributable to the high lithium ion mobility and the porous bimetallic nature of the cathode. To confirm that the incorporation of nickel impacted the overall device performance, the pure cobalt nanoflakes were used as the cathode.
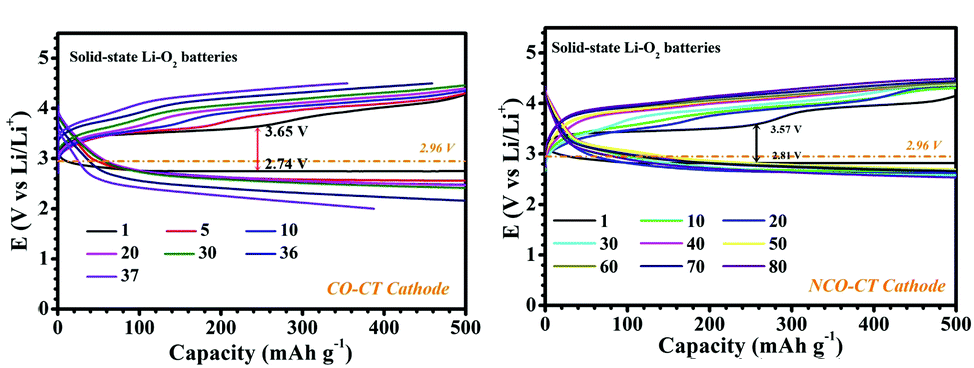
Figure 2. Cycling performance of cobalt (left) and cobalt-nickel cathodes (right) at a current density of 100 mA/g.
As seen in Figure 2, the cobalt-only batteries exhibit significant capacity loss in only 35 cycles whereas the NCO cathodes showed no degradation after 90 cycles. While cycling the NCO electrodes, the reversible formation of Li2O2, a common discharge product, occurred in the open pores of the cathode. These pores allow the 500 nm Li2O2 particles to form and dissolve without disrupting the structure of the cathode and give a more stable battery. This research brings completely solid-state lithium-oxygen batteries one step closer to reality.
To find out more, please read:
Hao Gong, Hairong Xue, Xueyi Lu, Bin Gao, Tao Wang, Jianping He and Renzhi Ma
Chem. Commun., 2019, 55, 10689-10692.
About the blogger:
 Beth Mundy is a PhD candidate in chemistry in the Cossairt lab at the University of Washington in Seattle, Washington. Her research focuses on developing new and better ways to synthesize nanomaterials for energy applications. She is often spotted knitting in seminars or with her nose in a good book. You can find her on Twitter at @BethMundySci.
Beth Mundy is a PhD candidate in chemistry in the Cossairt lab at the University of Washington in Seattle, Washington. Her research focuses on developing new and better ways to synthesize nanomaterials for energy applications. She is often spotted knitting in seminars or with her nose in a good book. You can find her on Twitter at @BethMundySci.








")
 Tianyu Liu obtained his Ph.D. (2017) in Chemistry from University of California, Santa Cruz in the United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at
Tianyu Liu obtained his Ph.D. (2017) in Chemistry from University of California, Santa Cruz in the United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at 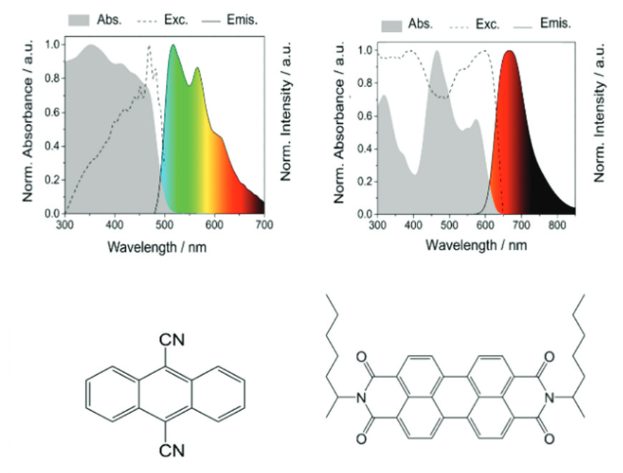






 Tianyu Liu obtained his Ph.D. (2017) in Chemistry from University of California, Santa Cruz in the United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at
Tianyu Liu obtained his Ph.D. (2017) in Chemistry from University of California, Santa Cruz in the United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at