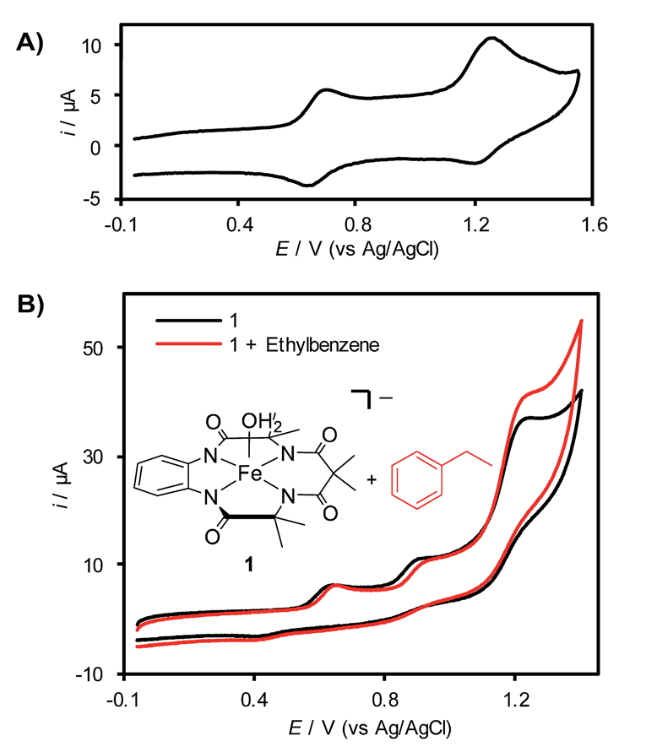
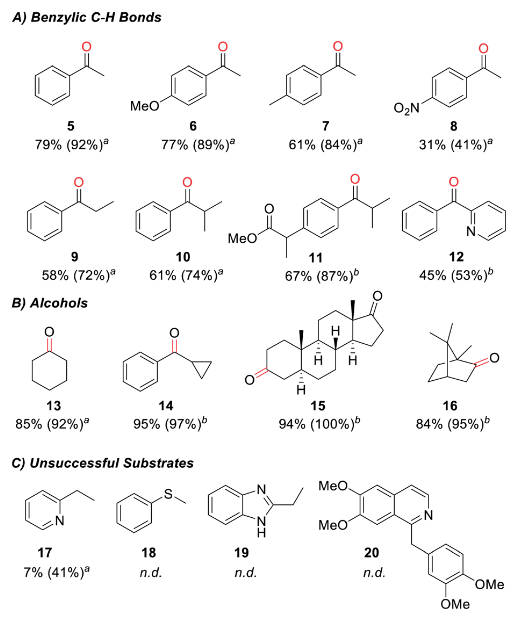
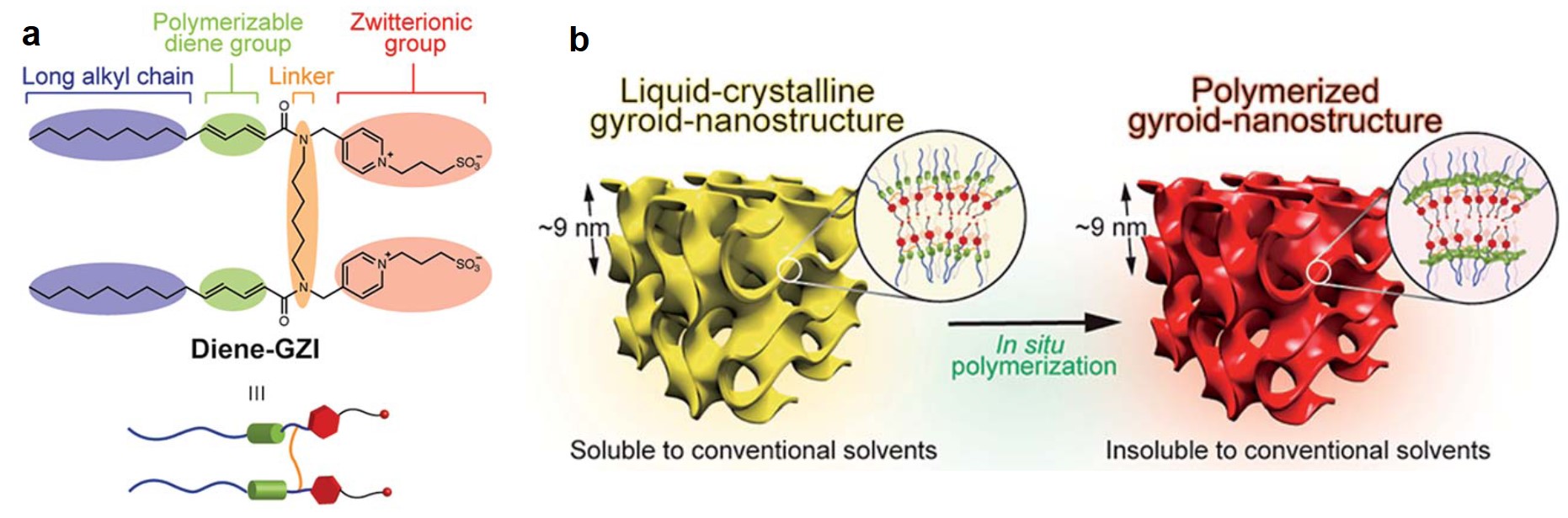
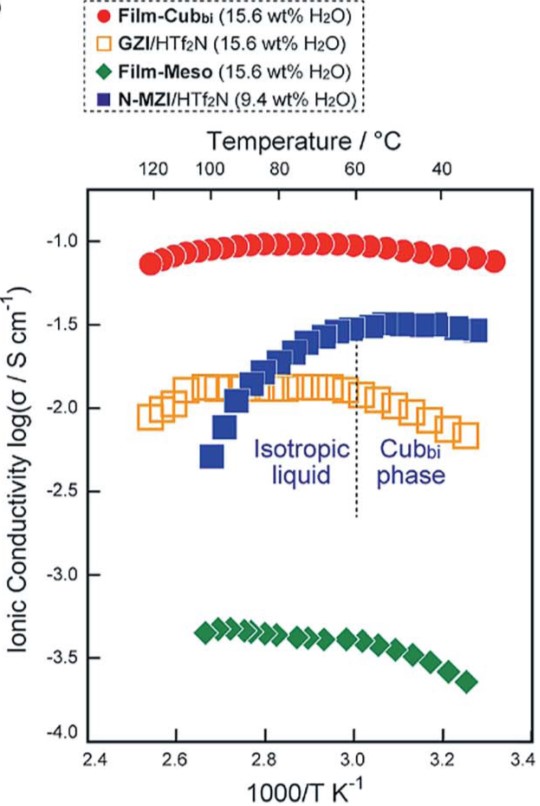
As a nanocrystal chemist, it pains me to say that sometimes nanoparticles aren’t enough. One strategy for engineering materials with complex functionalities is to embed nanoparticles into a larger host matrix structure. This has been widely studied for polymer-nanoparticle assemblies, but challenges abound for inorganic host matrices. Difficulties stem from problems controlling the microstructure of the host, as nanoparticles tend to accumulate at the borders of individual crystals in polycrystalline materials. Even when higher quality inorganic materials are made in the presence of nanocrystals loadings are below 1 wt%. A possible route to address these issues is by combining inorganic matrices with polymer-functionalized nanoparticles. The polymers can also form into vesicle structures that contain the nanoparticles which are larger and more compatible with characterization techniques.
One of those techniques is cryo-ptychographic X-ray computed tomography (cryo-PXCT), a fascinating and literally cool characterization method to image the internal structure of crystals. This is a variation on imaging techniques used heavily in medicine archeology to non-destructively visualize the interior of humans or artifacts. Cryo-PXCT cools the sample to -180 oC and has spatial resolution on the order of 50-70 nm. The researchers synthesized polymer vesicles and worms of approximately 232 nm in diameter and over 1 micron in length, respectively. The nanocomposites were made via an ammonia diffusion method with a solution of calcium chloride containing the polymer nano-structure exposed to gaseous ammonia and carbon dioxide to form CaCO3 crystals with nano-structure occlusions. The morphology of the nanocomposite crystal altered based on the type of occlusion – the vesicle/calcite combination retained a traditional calcite rhombohedral structure, while the worm/calcite composite crystals featured several rounded sides, an elongated shape, and only three flat faces. These composites possessed 15 – 25 wt% occlusions, significantly higher than prior work with pure nanoparticle incorporation.

Figure 1. SEM images of vessicle/calcite (left) and worm/calcite (right) nanocomposite single crystals.
Once prepared, the researchers examined the crystals by cryo-PXCT to determine the locations of the occlusions within the composites. In the vesicle/calcite composite the vesicles are non-uniformly distributed, with several layers of vesicle density, starting with a vesicle poor core, followed by a vesicle rich region, surrounded by another vesicle poor layer, with a slight vesicle enrichment near the surface. On average the vesicles are 300 nm apart and they maintain their shape, with the larger vesicles preferentially occluding in regions of higher occlusion densities.
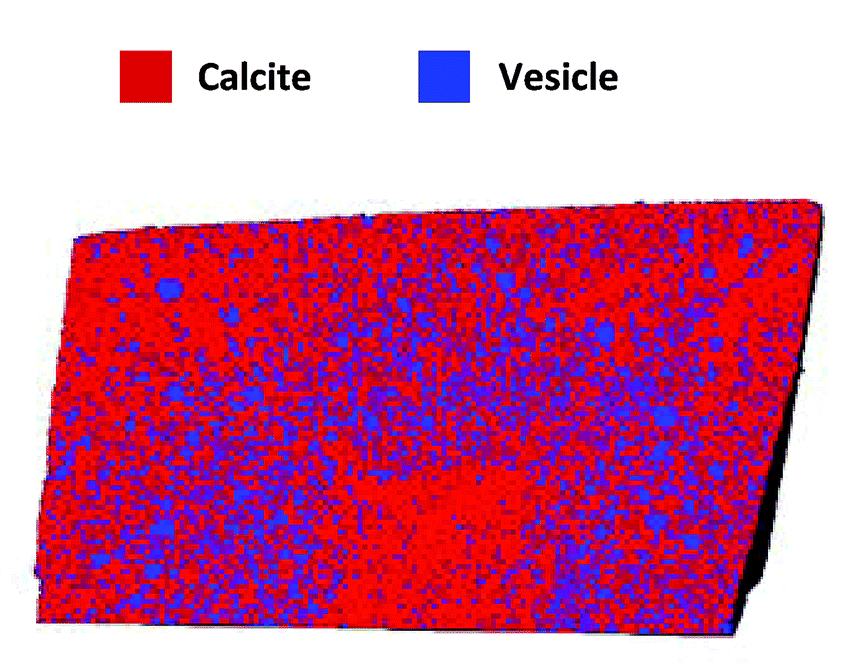
Figure 2. Rendering of slice through the vesicle/calcite nanocomposite colored to show both components.
The worm/calcite composite crystals show a very different distribution of occlusions, with an hourglass of low density in the center of the crystal, surrounded by a worm rich zone, and an exterior worm poor layer. These zoning effects are likely determined by the interactions between the polymers and the growing crystal surfaces or the calcium cations in the solution. Cryo-PXCT offers a fascinating way to probe the internal structure of novel multicomponent crystals in three dimensions with nanoscale resolution, providing valuable information to eventually help determine structure-function relationships.
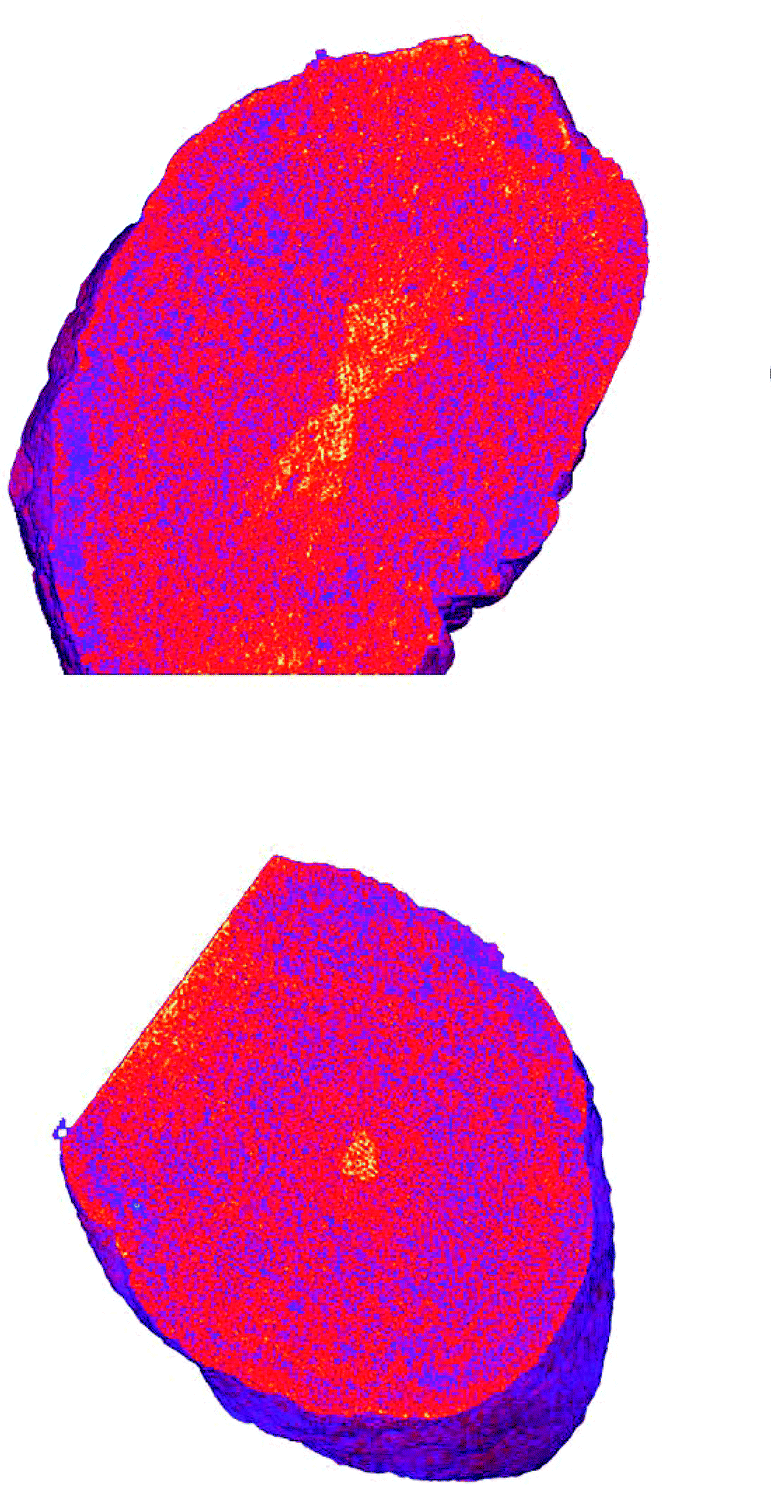
Figure 3. Tomographs of worm/calcite nanocomposites showing the localization of worms in an hourglass shape in the center of the crystal.
To find out more, please read:
Ptychographic X-ray tomography reveals additive zoning in nanocomposite single crystals
Johannes Ihli, Mark A. Levenstein, Yi-Yeoun Kim, Klaus Wakonig, Yin Ning, Aikaterini Tatani, Alexander N. Kulak, David C. Green, Mirko Holler, Steven P. Armes and Fiona C. Meldrum
Chem. Sci., 2020, 11, 355-363.
About the blogger:
 Beth Mundy is a PhD candidate in chemistry in the Cossairt lab at the University of Washington in Seattle, Washington. Her research focuses on developing new and better ways to synthesize nanomaterials for energy applications. She is often spotted knitting in seminars or with her nose in a good book. You can find her on Twitter at @BethMundySci.
Beth Mundy is a PhD candidate in chemistry in the Cossairt lab at the University of Washington in Seattle, Washington. Her research focuses on developing new and better ways to synthesize nanomaterials for energy applications. She is often spotted knitting in seminars or with her nose in a good book. You can find her on Twitter at @BethMundySci.








")
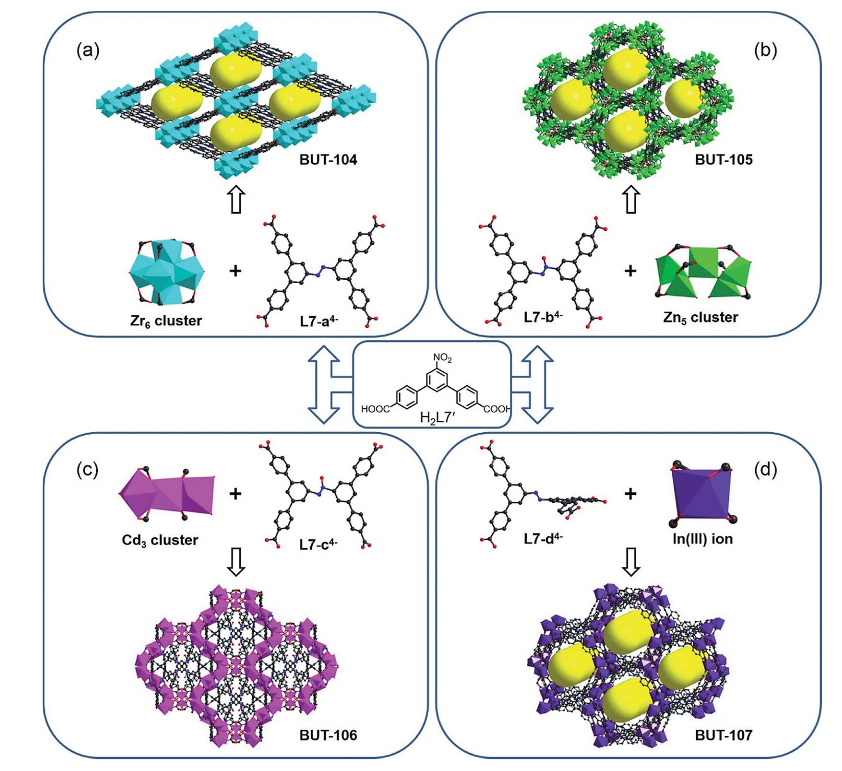
 Tianyu Liu obtained his Ph.D. (2017) in Chemistry from University of California, Santa Cruz in the United States. He is passionate about the communication of scientific endeavors and cutting-edge research to both the general public and other scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at
Tianyu Liu obtained his Ph.D. (2017) in Chemistry from University of California, Santa Cruz in the United States. He is passionate about the communication of scientific endeavors and cutting-edge research to both the general public and other scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at