Australian scientists from The University of Adelaide and Graz University of Technology recently made a breakthrough in protein chemistry. They revealed that the key to successfully encasing proteins into metal organic frameworks (MOFs, a family of highly porous organic-metal coordination molecules) was the surface charge of the proteins. Their work was recently published in Chemical Science.
Encapsulating proteins into MOFs, a process termed “biomimetic mineralisation”, is an efficient way to protect and preserve proteins. This process is typically initiated by mixing the proteins and the precursors of a MOF. The MOF starts to grow at the protein surface and eventually fully covers the protein. As the growth of the MOF begins with nucleation on the protein surface, the surface properties play an important role in controlling the rate and quality of the encapsulation. Unfortunately, the interplay between protein surface and MOF growth has not been well understood, leading to inefficient reactions that require excess MOF precursors and long reaction times.
The authors demonstrated for the first time that the surface charge is one of the key factors that affects the possibility of biomimetic mineralisation. Specifically, they discovered that proteins with strongly negative charged surfaces, such as pepsin and bovine serum albumin, were able to be spontaneously incorporated into ZIF-8 (a benchmark MOF). Conversely, those with naturally positively charged or slightly negatively charged surfaces, including haemoglobin, were incapable of forming composites with ZIF-8. The authors further showed that changing the surface charge could allow or prohibit the encapsulation. For example, after reacting the lysine groups of haemoglobin with succinic anhydride, the surface of haemoglobin became more negative and ZIF-8 could now readily wrap around the protein (Figure 1). The surface potential threshold to induce biomimetic mineralisation was determined to be -30 mV.
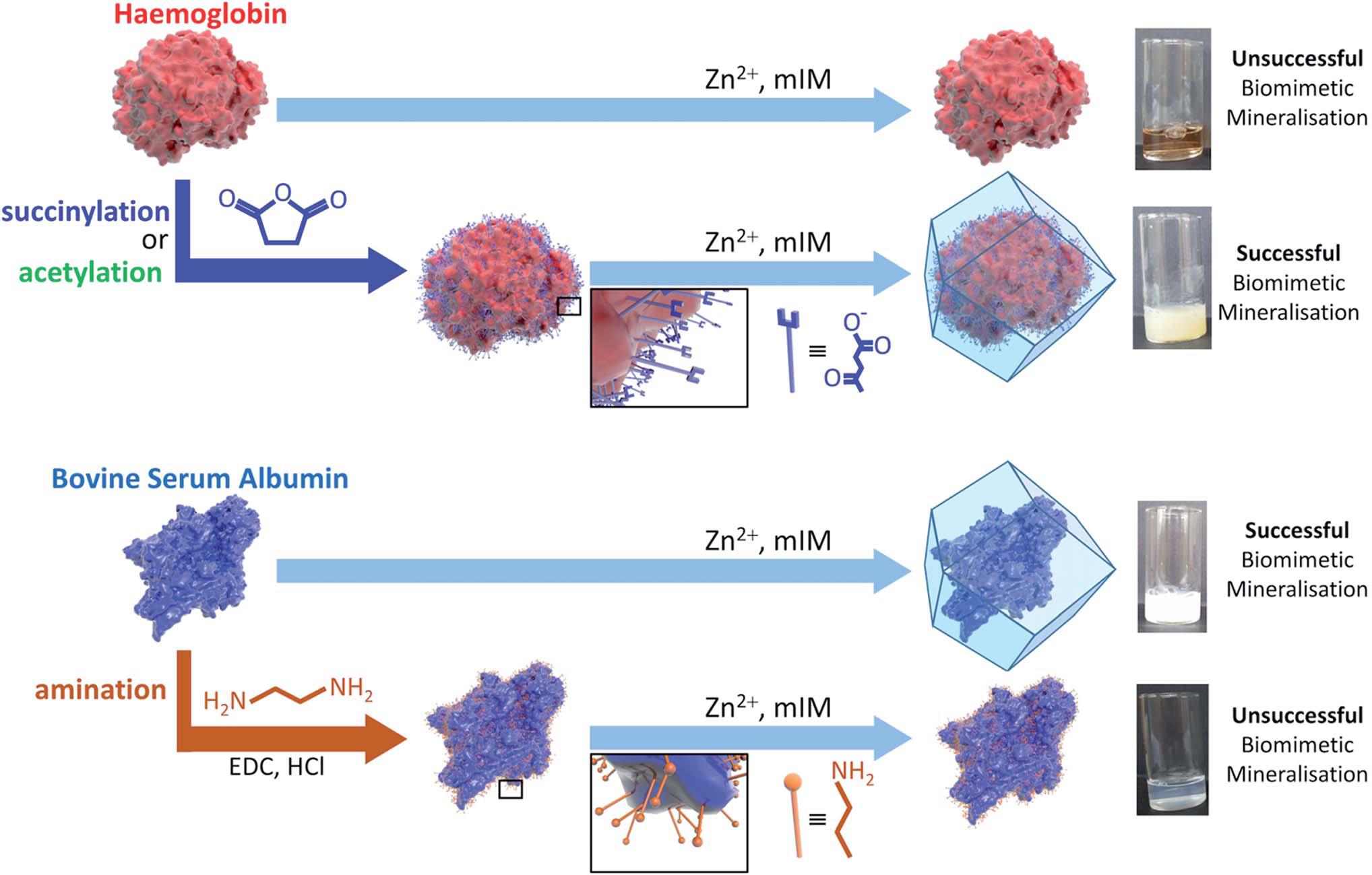
Figure 1. Schematic illustrations of the biomimetic mineralisation of haemoglobin and bovine serum albumin. Haemoglobin with slightly negatively charged surface is unable to form composites with ZIF-8, but becomes active after succinylation or acetylation to make its surface strongly negatively charged. On the contrary, bovine serum albumin with a strongly negatively charged surface readily combines with ZIF-8, but loses its activity when its surface becomes less negatively charged via amination.
The mechanism of the aforementioned observations was attributed to the electrostatic attraction between the protein surface and Zn2+, one of the MOF precursors. The more negatively the surface is charged, the more easily the Zn2+ will attach to and accumulate at the protein surface. The adsorbed Zn2+ ions then serve as the nucleation sites for the MOF to grow around the protein. This hypothesis is proven by a number of control experiments and also validated by computational studies.
This study highlights the surface potential of a protein as a critical factor in its ability to induce biomimetic mineralisation with MOFs. The conclusions could potentially be extended to biomolecules other than proteins (e.g. viruses and cells) to facilitate their integration with various MOFs.
To find out more please read:
Natasha K. Maddigan, Andrew Tarzia, David M. Huang, Christopher J. Sumby, Stephen G. Bell, Paolo Falcaro and Christian J. Doonan
Chem. Sci., 2018 , DOI: 10.1039/c8sc00825f
About the blogger:
 Tianyu Liu obtained his Ph.D. (2017) in Physical Chemistry from University of California, Santa Cruz in United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at http://liutianyuresearch.weebly.com/.
Tianyu Liu obtained his Ph.D. (2017) in Physical Chemistry from University of California, Santa Cruz in United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at http://liutianyuresearch.weebly.com/.








")