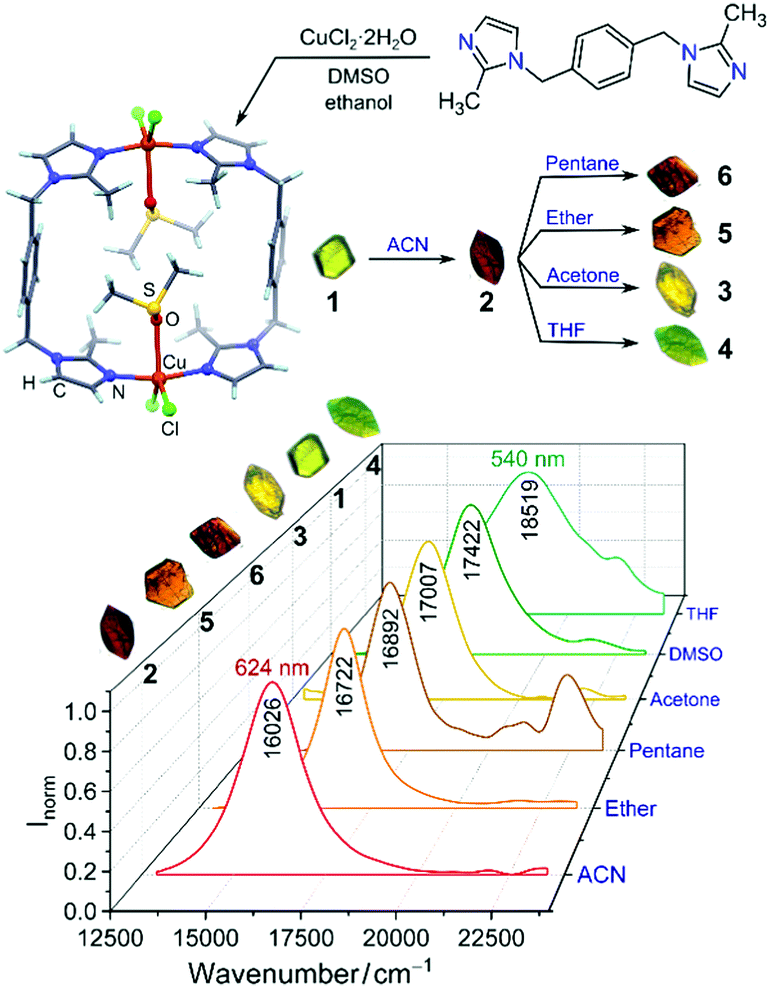
Synthesis of copper bimetallic complexes from imidazolyl ligands, and the solvatochromic materials formed upon crystallization and solvent guest-exchange. The solvatochromic behaviour was quantified with visible-region diffuse reflectance spectra.
During the first inorganic chemistry course I took during my undergraduate degree, our professor started the class by passing around some mineral samples, promising us that if we pursued the chemistry of metals we could work with beautifully coloured crystals every day. At the time, colour seemed like such a trite detail amongst the complexity of the subject. Why would you choose a field of study based on something so simple? Well, after a PhD dominated by pale yellow oils, I think I get it now.
Nikolayenko and Barbour at the University of Stellenbosch in South Africa bring us colour! The authors synthesised organometallic copper complexes, which crystallise to form porous single crystals that drastically change colour upon absorption of various solvents. The authors investigated the solvatochromic mechanism using X-ray crystallography, EPR, UV-visible spectroscopy and DFT calculations. Solvatochromic materials are not just made to look pretty; they have potential to be used as sensitive, selective and recyclable sensors to detect solvent vapours with useful applications in industrial process risk management, chemical threat detection and environmental monitoring.
The researchers synthesised a series of complexes comprised of a bidentate ligand with 2-methylimidazolyl groups coordinated to copper(II) ions. The complexes stack to form channels in the crystal, capable of trapping solvent molecules to give different coloured crystals: DMSO and THF-containing crystals are green (λmax = 574 nm and 540 nm, respectively), those containing acetonitrile are red (λmax = 624 nm), and crystals trapping acetone, ether and pentane are yellow (λmax = 588), orange (λmax = 598 nm) and red/brown (λmax = 592 nm), respectively.
The authors revealed a correlation between the size of the solvent guest, coordination geometry of the copper complex, and the ligand field splitting. Small guests such as acetonitrile minimally perturb the metallocyclic framework, preserving a rhombic ligand field geometry (large δxy of g values in the EPR spectrum), small ligand d-orbital splitting and red-shifted optical spectra. Large guests such as THF have the opposite effect, giving ligand field geometries approaching tetragonal (small δxy), large ligand field d-orbital splitting and blue-shifted optical spectra.
By delving into the complexity beneath a seemingly simple phenomenon, Nikolayenko, Barbour and their co-workers have shown using a series of single-crystal complexes that there is nothing simple about colour (and nothing trite about detail).
To find out more please read:
Supramolecular solvatochromism: mechanistic insight from crystallography, spectroscopy, and theory
Varvara I. Nikolayenko, Lisa M. van Wyk, Orde Q. Munro, Leonard J. Barbour.
Chem. Commun., 2018, Advance Article
DOI: 10.1039/c8cc02197j

About the author
Zoë Hearne is a PhD candidate in chemistry at McGill University in Montréal, Canada, under the supervision of Professor Chao-Jun Li. She hails from Canberra, Australia, where she completed her undergraduate degree. Her current research focuses on transition metal catalysis to effect novel transformations, and out of the lab she is an enthusiastic chemistry tutor and science communicator.








")
 Tianyu Liu obtained his Ph.D. (2017) in Physical Chemistry from University of California, Santa Cruz in the United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at
Tianyu Liu obtained his Ph.D. (2017) in Physical Chemistry from University of California, Santa Cruz in the United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at 
![Synthesis of oxazolines from ketones and isocyanaoacetate esters via a formal [3+2] cycloaddition reaction catalysed by a multicatalytic system of silver and a dihydroquinine squaramide organocatalyst](https://blogs.rsc.org/cc/files/2018/04/Screenshot-2018-04-03-16.24.41.png)

 Dr. Rafal Klajn
Dr. Rafal Klajn