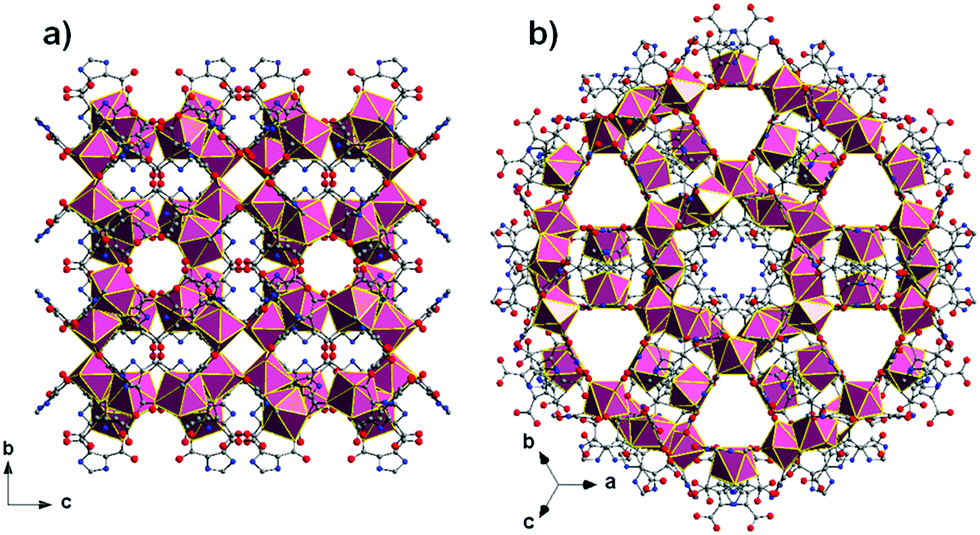
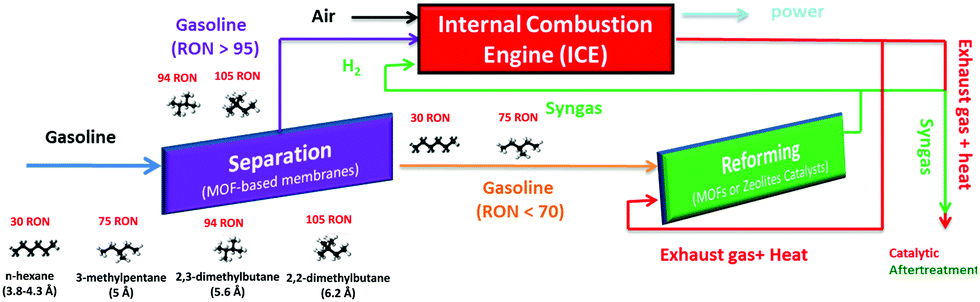
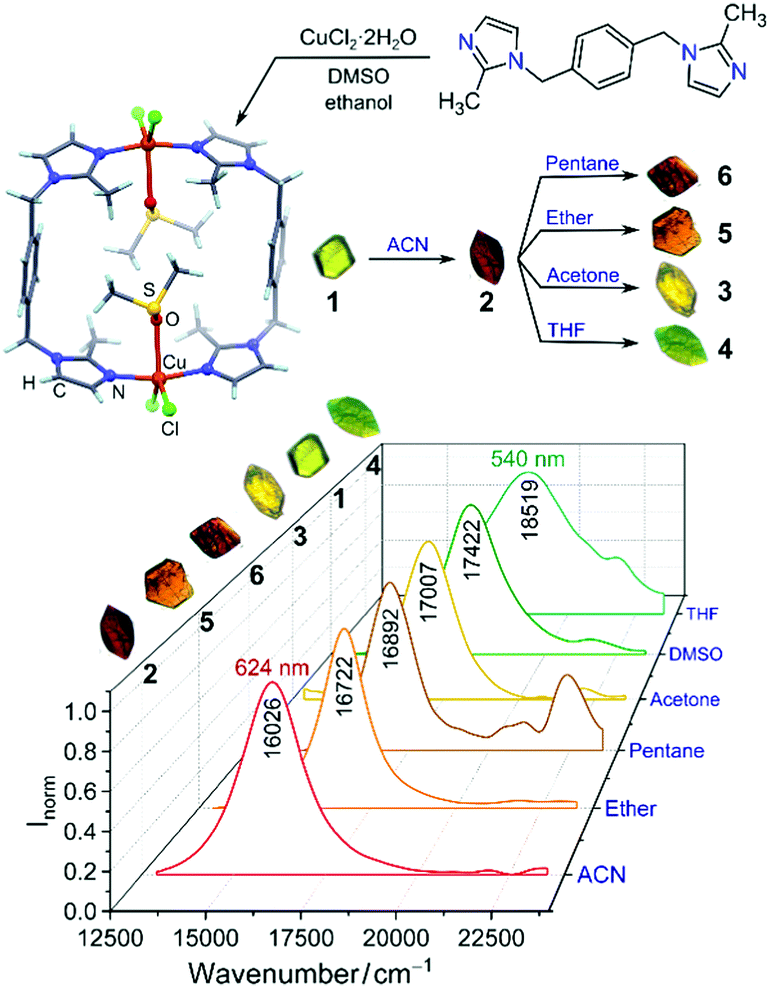
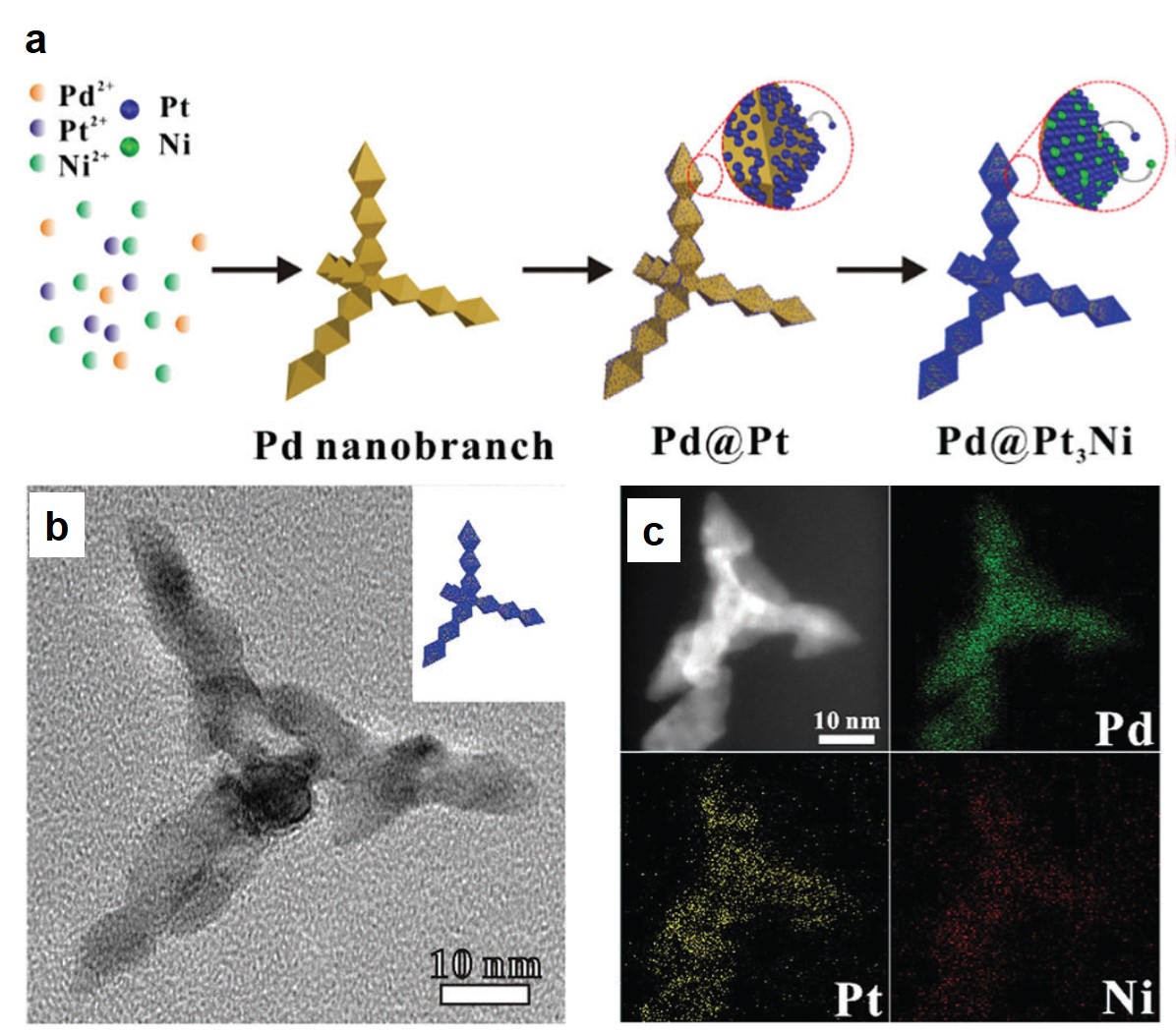
Increasing the volumetric energy densities of batteries is essential for improving the durability of portable electronics and the operating ranges of electric vehicles. One way to improve energy density is to enlarge the mass fraction of active materials in battery electrodes; however, the degree of enhancement remains limited. This limitation results from the densification of the electrodes when the mass fraction increases, making electron transport and ion diffusion throughout the electrodes sluggish. These drawbacks lower the utilization efficiency of the overall electrode materials.
A team of scientists from China and the United States has recently addressed the aforementioned challenges. Specifically, they synthesized a 3D cathode of carbon-coated Li2MnSiO4 (Li2MnSiO4/C) with a structure mimicking a honeycomb filled with bee pupas (Fig. 1). This lithium-ion battery cathode possesses a high mass fraction of 90% (of overall electrode mass) as well as a volumetric energy density as high as 2443 Wh/dm3.
The uniquely structured electrodes were prepared through a hard-template method (Fig. 1). Using polystyrene particles, silica surface coating, and Li2MnSiO4 precursor infiltration, the authors synthesized a carbon-coated Li2MnSiO4 honeycomb scaffold with each cavity filled with a carbon-coated Li2MnSiO4 particle. This architecture differed from previously reported 3D structures, which typically had a large portion of voids, and enabled an ultrahigh active-material mass loading of 90 wt.%. Additionally, the gaps between the scaffold and the particles functioned as ion-diffusion channels, and the carbon coatings served as electron-transport expressways. These characteristics effectively addressed the problem of sluggish ion diffusion and electron transport.

Figure 1. The synthesis procedures of the BPFH-shaped Li2MnSiO4/C electrode. The green particles and yellow scaffold represent polystyrene spheres and the silica coating, respectively.
Due to the facilitated electron transport and ion diffusion, the Li2MnSiO4/C electrode with a bee pupa-filled honeycomb (BPFH) structure (Fig. 2a) exhibited an outstanding charge-storage performance. Specifically, it delivered a high volumetric capacity of 643 mAh/cm3 at a current density of 0.1 C, corresponding to a volumetric density of 2443 Wh/dm3. This volumetric capacity was approximately two times higher than that of a Li2MnSiO4/C honeycomb lattice without any Li2MnSiO4 particles (Fig. 2b). After 100 consecutive charge-discharge cycles, the BPFH-shaped Li2MnSiO4/C electrode retained a volumetric capacity of 328 mAh/cm3 (Fig. 2c).
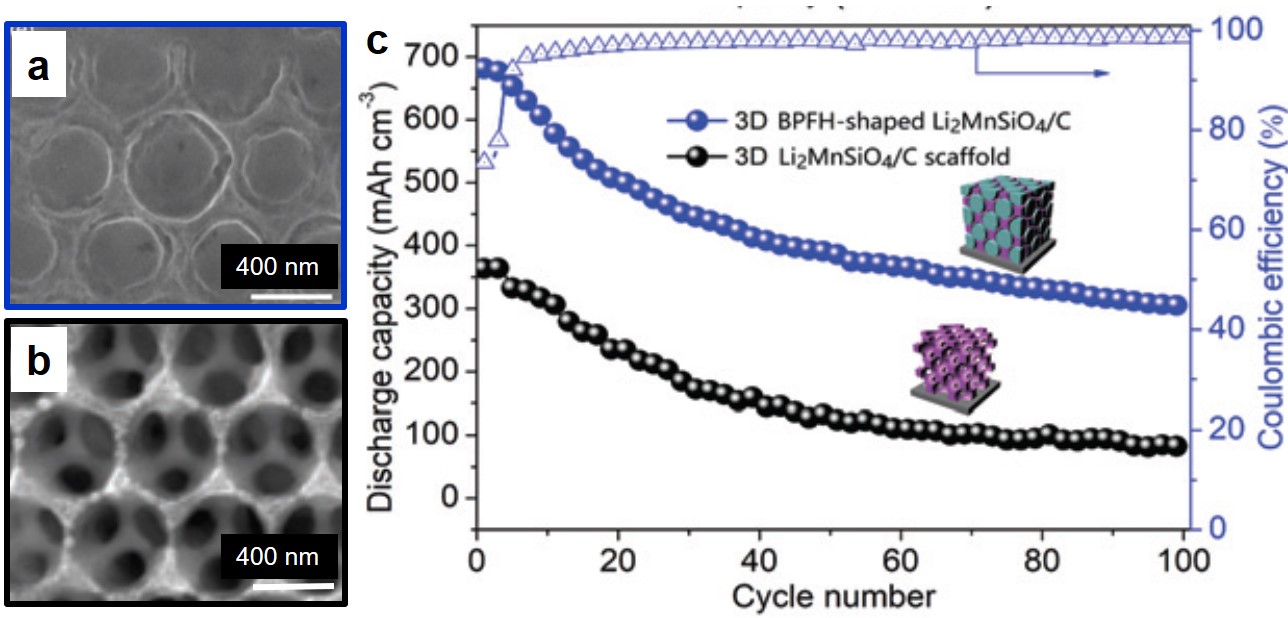
Figure 2. (a and b) Scanning electron microscopy images of (a) the BPFH-shaped Li2MnSiO4/C electrode and (b) the Li2MnSiO4/C scaffold. (c) The capacities and the Coulombic efficiencies of the two electrodes during 100 charge-discharge cycles.
The demonstrated BPFH architecture could be extended to other materials for the synthesis of battery electrodes with both high mass fractions of active materials and outstanding volumetric energy densities.
To find out more please read:
Jinyun Liu, Xirong Lin, Huigang Zhang, Zihan Shen, Qianqian Lu, Junjie Niu, Jinjin Li and Paul V. Braun
Chem. Commun., 2019, 55, 3582-3585
About the blogger:
 Tianyu Liu obtained his Ph.D. (2017) in Chemistry from University of California, Santa Cruz in the United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at http://liutianyuresearch.weebly.com/.
Tianyu Liu obtained his Ph.D. (2017) in Chemistry from University of California, Santa Cruz in the United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at http://liutianyuresearch.weebly.com/.








")
 About the author
About the author
 Tianyu Liu obtained his Ph.D. (2017) in Chemistry from University of California, Santa Cruz in the United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at
Tianyu Liu obtained his Ph.D. (2017) in Chemistry from University of California, Santa Cruz in the United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at