Activating a bond is the first step towards bond breaking processes for synthesis and catalysis. Despite the major role of transition metals in a variety of bond activation processes, C–X bond activation of halogenated benzenes (PhX, X = F, Cl, Br, I) is still challenging; there are very few examples of metal···XPh complexes, even though they are crucial for C–X bond activation and catalysis. There are even fewer examples of main group metal···XPh complexes, with no examples of main group complexes of bromobenzene or iodobenzene.
Researchers in Germany have been studying cationic magnesium complexes with the β-diketiminate ligand (RBDI, R = methyl or t-butyl), where their extreme Lewis acidity makes them ideal candidates for halobenzene complex formation for C–X activation. Building upon their previous findings of the formation of a Mg-chlorobenzene complex, the researchers have now demonstrated the preparation of the full series of halobenzene complexes, including the first examples of coordination of bromobenzene and iodobenzene to a main group metal, as shown in Scheme 1.
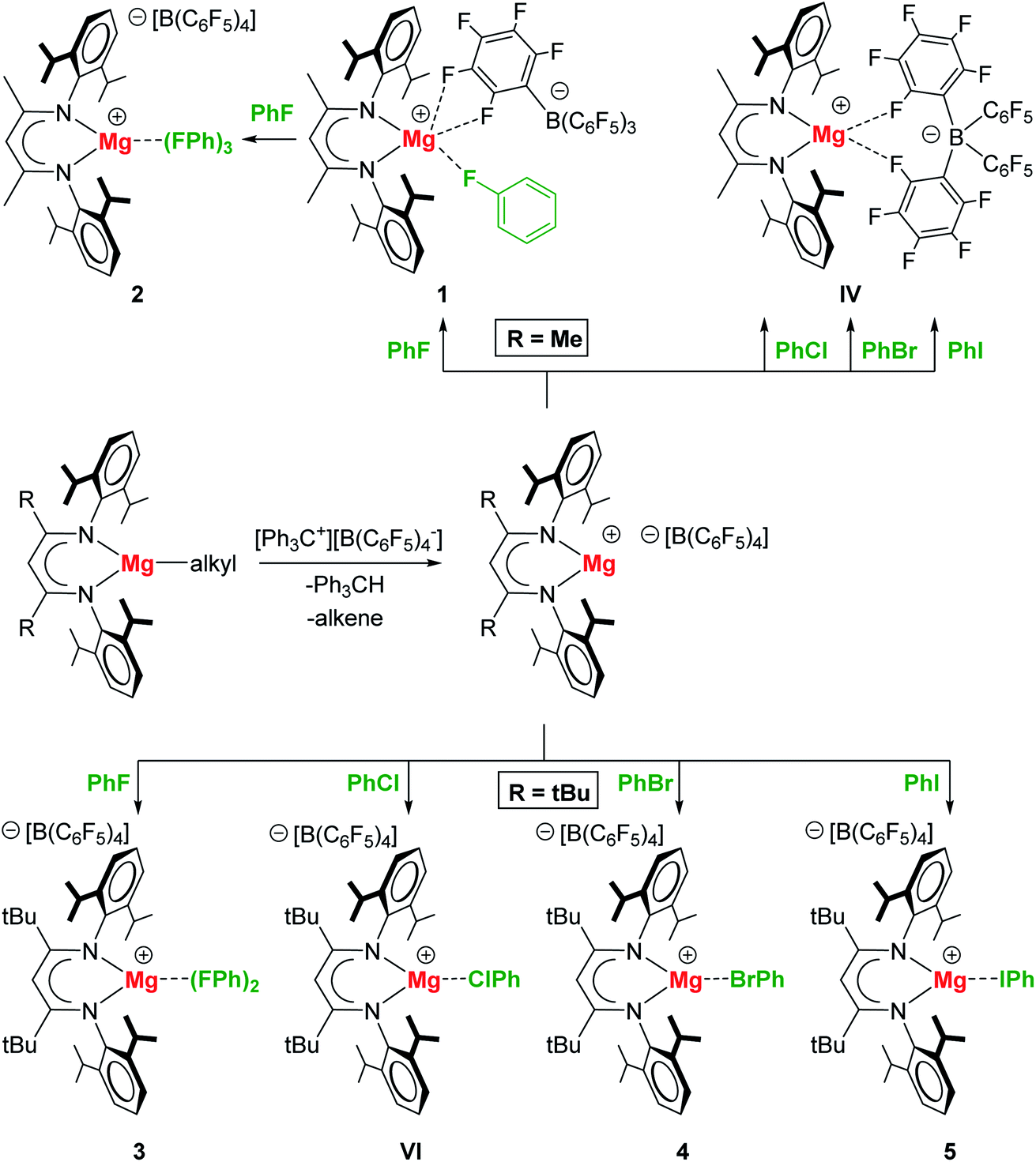
Scheme 1. Syntheses of Mg-XPh complexes (X = F, Cl, Br, I)
The researchers found that both the smaller methyl-substituted complex, (MeBDI)Mg+, and the bulkier t-butyl substituted complex, (tBuBDI)Mg+, were able to bind fluorobenzene to form Mg···FPh complexes (1–3 in, Scheme 1), owing to the high polarity of PhF that can compete with the Mg···B(C6F5)4– (the magnesium–anion) interaction. The other halobenzene complexes (Mg···XPh for X = Cl, Br, I; VI, 4, 5) could only be accessed with the use of the bulkier tBuBDI ligand. This was attributed to the fact that the bulky t-butyl substituents essentially turn off the Mg···B(C6F5)4– interaction, which in turn allows the less polar PhX halobenzenes to bind to the magnesium centre.
The researchers isolated and fully characterised the Mg-halobenzene complexes, and used X-ray crystallography and DFT calculations to further understand their properties. The interaction of the strongly Lewis acidic (BDI)Mg+ cation with the halobenzene resulted in C–X activation as shown by elongation of the C–X bonds in the crystal structures. Additionally, the solid-state structures showed that the Mg···X–Ph angle is the most linear for PhF and decreases in size (i.e. bends more) for the larger halogens. This increased bending for the larger halogens is explained by the halogen σ-hole, which is a region of positive electrostatic potential on the surface of the halogen opposite to the C–X bond, that increases with halogen size. As shown by the schematic in Figure 1, the presence of a larger halogen hole forces a more acute Mg···X interaction relative to the C–X bond.
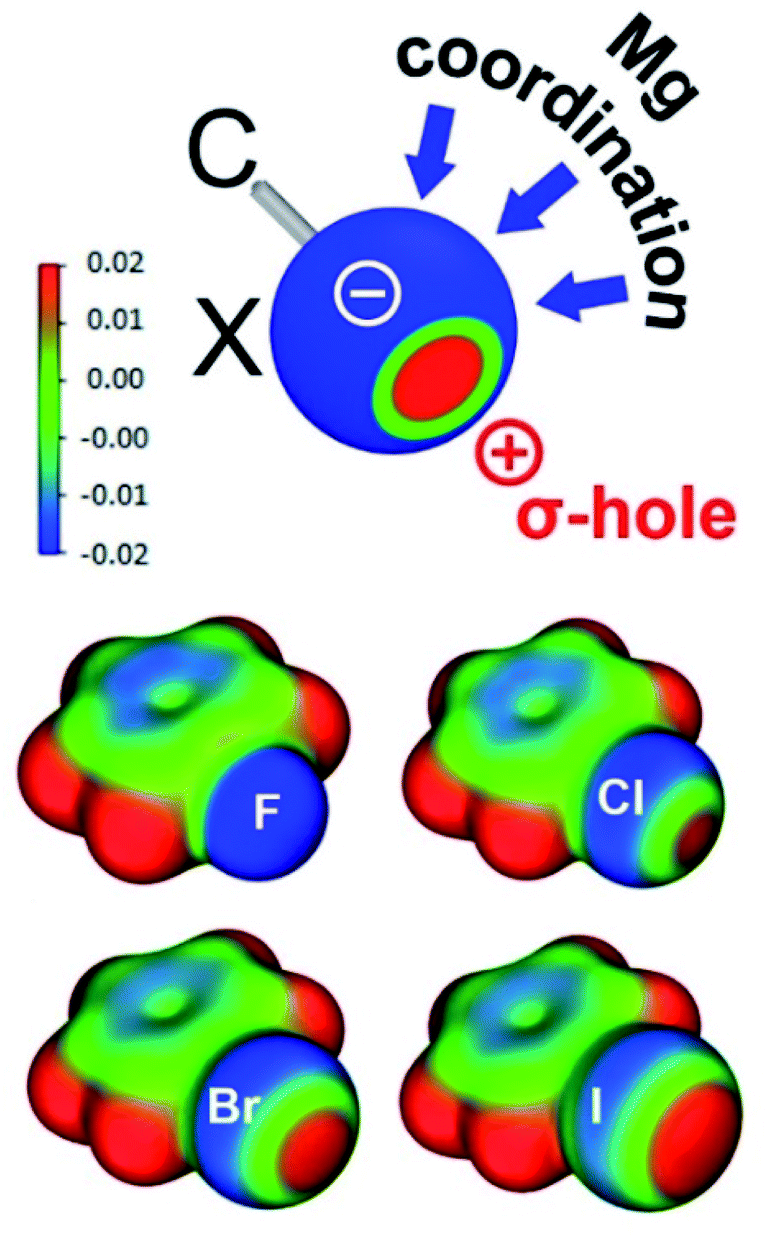
Figure 1. Top: Schematic showing the halogen σ-hole (red = positive electrostatic potential, blue = negative electrostatic potential), with possible coordination sites for Mg. Bottom: Electrostatic maps for the halobenzenes.
DFT calculations were also performed and were in good agreement with the solid-state experimental parameters. The researchers calculated complexation enthalpies between 11 and 13 kcal mol-1, which are weak but still indicate a Mg···X–Ph interaction. This interaction ultimately indicates C–X bond activation, signifying that these main group complexes show potential for C–X bond breaking processes in future catalytic applications.
To find out more, please read:
Magnesium–halobenzene bonding: mapping the halogen sigma-hole with a Lewis-acidic complex
Alexander Friedrich, Jürgen Pahl, Jonathan Eyselein, Jens Langer, Nico van Eikema Hommes, Andreas Görling and Sjoerd Harder*
Chem. Sci., 2021, Advance Article
About the blogger:
 Dr. Samantha Apps recently finished her post as a Postdoctoral Research Associate in the Lu Lab at the University of Minnesota, USA, and obtained her PhD in 2019 from Imperial College London, UK. She has spent the last few years, both in her PhD and postdoc, researching synthetic nitrogen fixation and transition metal complexes that can activate and functionalise dinitrogen. Outside of the lab, you’ll likely find her baking at home, where her years of synthetic lab training has sparked a passion in kitchen chemistry too.
Dr. Samantha Apps recently finished her post as a Postdoctoral Research Associate in the Lu Lab at the University of Minnesota, USA, and obtained her PhD in 2019 from Imperial College London, UK. She has spent the last few years, both in her PhD and postdoc, researching synthetic nitrogen fixation and transition metal complexes that can activate and functionalise dinitrogen. Outside of the lab, you’ll likely find her baking at home, where her years of synthetic lab training has sparked a passion in kitchen chemistry too.








")