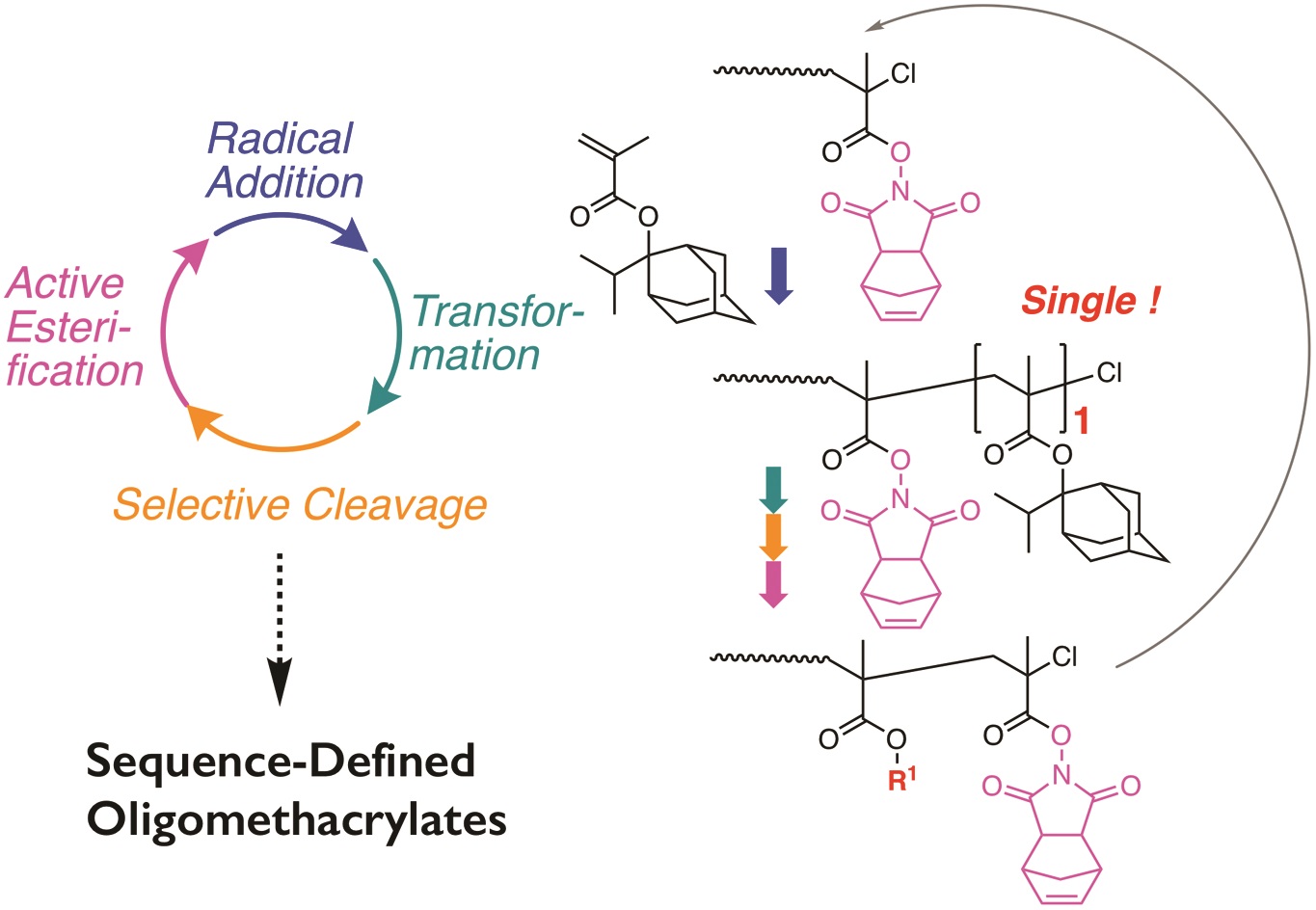
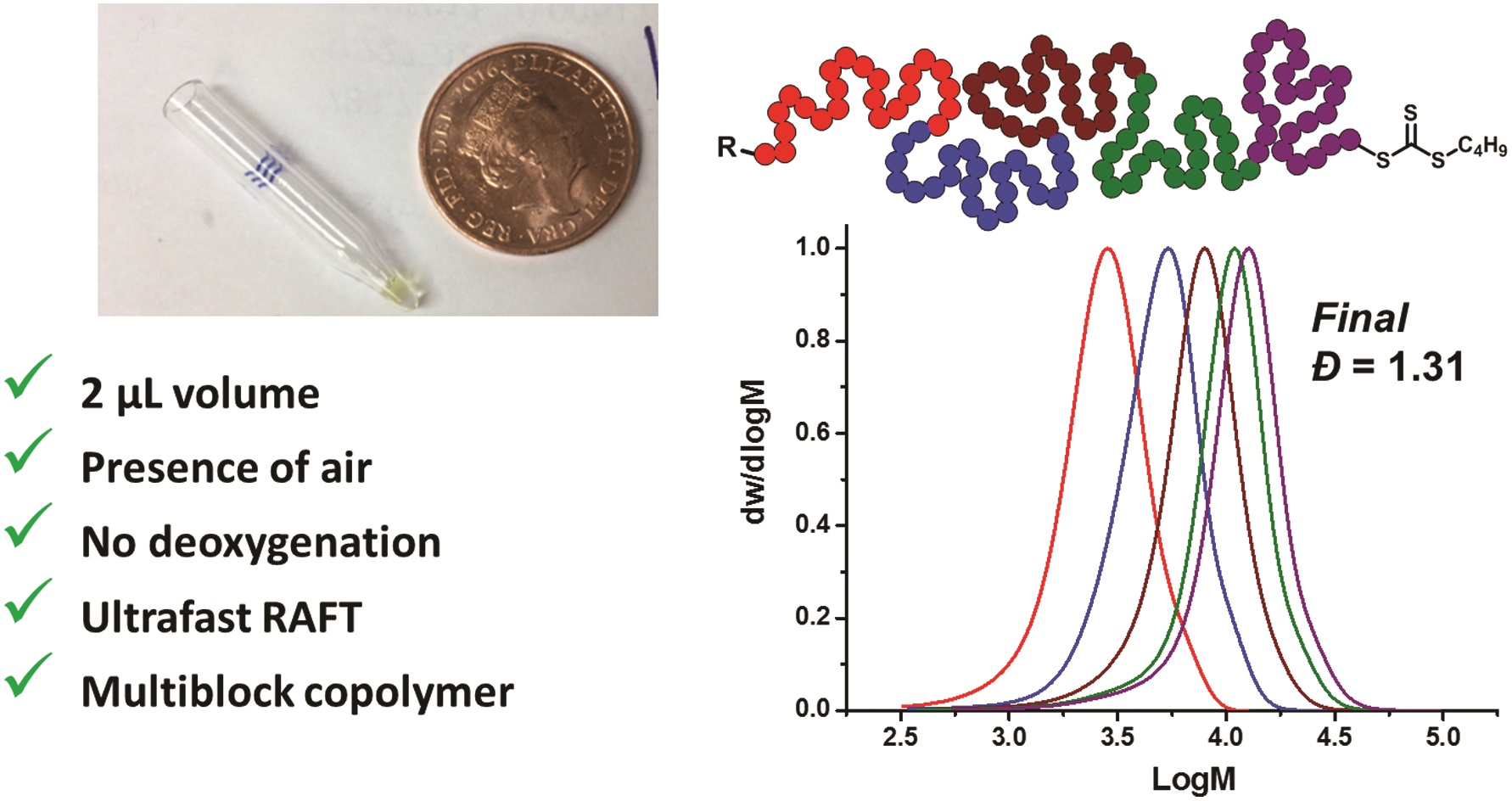
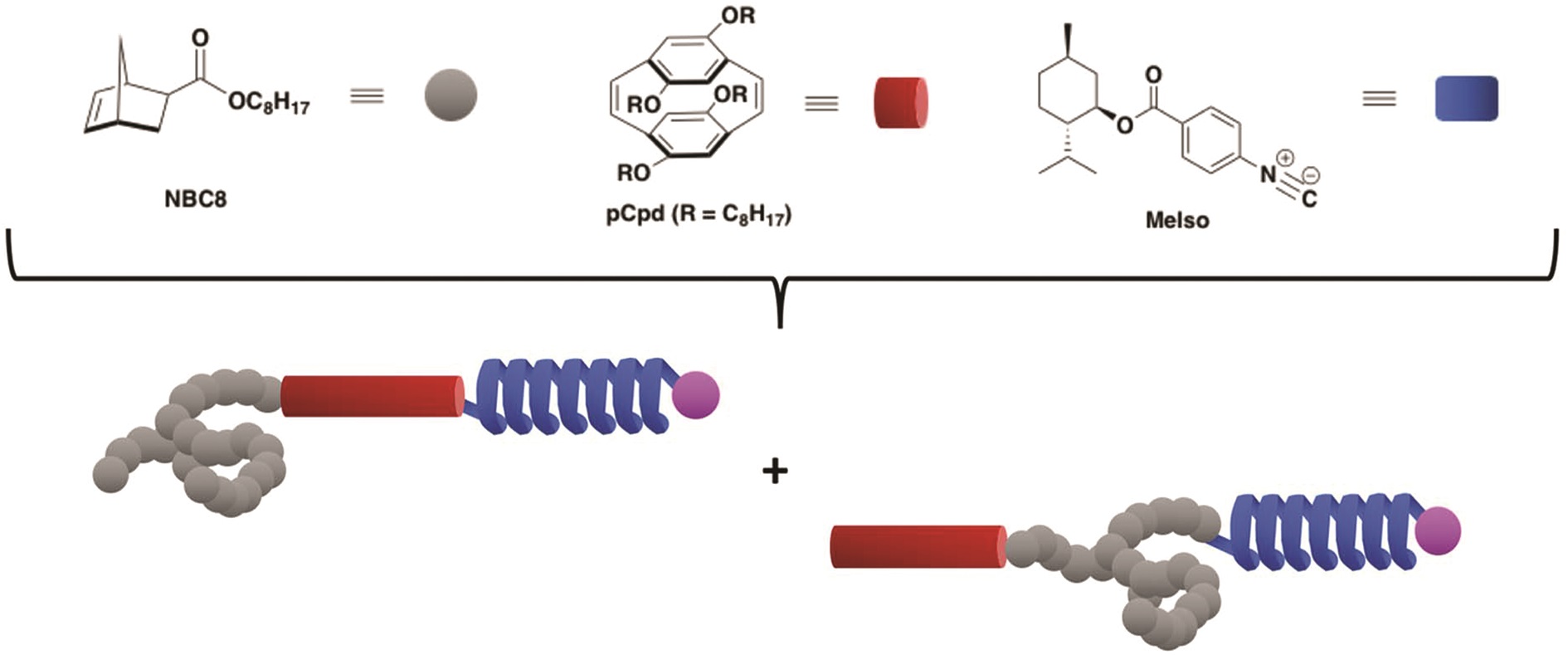
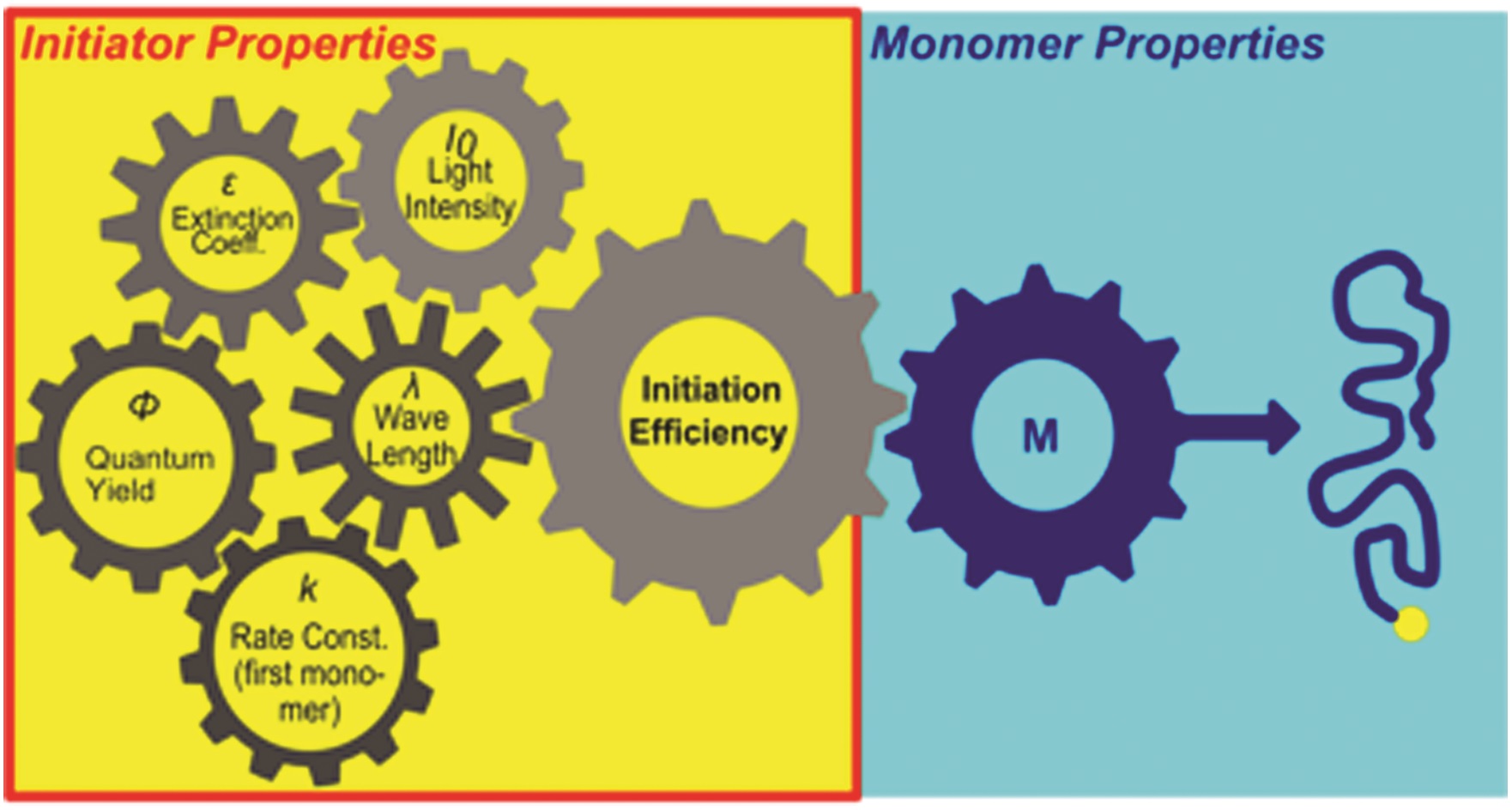
Nanotransfer printing is a technique often used to construct complex patterns by employing elastomeric stamps and relying on surface chemistries. This has enabled not only the assembly of complex constructs but also the effective integration of heterogeneous materials. In the current manuscript, Campos and co-workers significantly contributed to this direction by introducing a nanotransfer technique, termed soft pattern-transfer printing, which does not rely on adhesive layers or external stimuli. As a result, a cost and time efficient high throughput processing platform is being developed. To achieve this, representative organic thin films of P3HT homopolymers, self-assembled diblock copolymers and functionalized perylene diimide small molecules were employed as inks for micron-sized array of patterns ranging from squares, lines, polygons and rings. Importantly, hierarchical patterns were obtained through microns-sized arrays of self-assembled block copolymers. In addition, to build layers of complex structures onto the same film, the technique can be repeated through sequential printing. As the authors elude in their conclusion, such high-fidelity pattern transfer work is very promising for potential uses in a number of areas such as the construction of van der Waals heterostructures interfaced with self-assembled block copolymer thin films and the development of platforms to investigate the influence of hierarchical patterning on cell differentiation.

Tips/comments directly from the authors:
- Solvent-vapor induced self-assembly of diblock copolymer thin films is an attractive approach to achieve long-range microphase segregation.
- Achieving solvent-vapor induced self-assembly of diblock copolymer thin films directly on exfoliated materials is particularly challenging because of the macroscopic topographical heterogeneities which disrupt the film integrity.
- Moreover, the generation of hierarchical patterns, particularly with one length scale in the nanometer regime, often involves lithographic processes which are difficult to scale.
- A simple contact-based approach is presented for transfer of polymeric materials (e.g. self-assembled block copolymers, homopolymers, small molecules), with well-defined edge resolution (<20 nm) and high fidelity of nanoscale pattern transfers.
- To avoid warped or cracked transfers, it is critical to handle PDMS stamps with care, avoiding excessive mechanical deformation, and to apply minimal pressure.
- Importantly, we show successful transfer of solvent-vapor induced self-assembled diblock copolymer films onto 2D materials (e.g. boron nitride).
- The transfer of micron-scale patterns of self-assembled diblock copolymers with nanoscale features yield hierarchical ordering.
- Patterns resulting from sequential soft nanotransfer printing resemble Moiré patterns, large-scale interference patterns. Such complex patterns may be used to impart local physical and electronic perturbations.
Read this article for free until the 31st July!
Hierarchical patterns with sub-20 nm pattern fidelity via block copolymer self-assembly and soft nanotransfer printing, Polym. Chem., 2019, 10, 3194-3200, DOI: 10.1039/C9PY00335E
About the Web writer
 Dr. Athina Anastasaki is an Editorial Board Member and a Web Writer for Polymer Chemistry. Since January 2019, she joined the Materials Department of ETH Zurich as an Assistant Professor to establish her independent research group.
Dr. Athina Anastasaki is an Editorial Board Member and a Web Writer for Polymer Chemistry. Since January 2019, she joined the Materials Department of ETH Zurich as an Assistant Professor to establish her independent research group.








")
![c9py00213h-ga[1]](https://blogs.rsc.org/py/files/2019/06/c9py00213h-ga1.jpg)