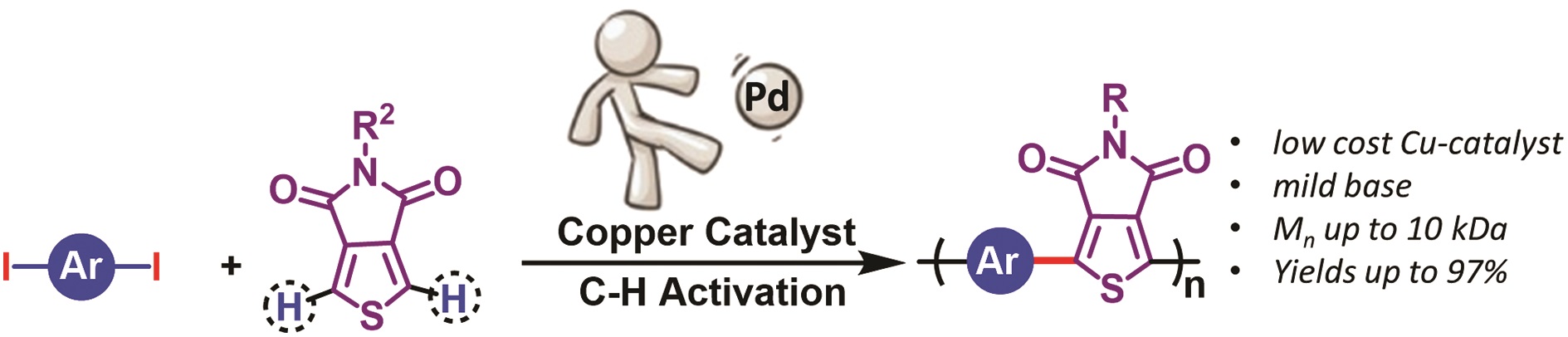
Direct arylation polymerization is a useful methodology that allows for the preparation of conjugated polymers. One of the great advantages of this technique is that it eliminates the use of toxic and pyrophoric reagents typically utilized to prepare monomers for other polymerization methods (e.g. Stille, Suzuki, etc.). Importantly, the technique allows the preparation of polymers with undetectable levels of homo-coupling or branching defects leading to the defect free synthesis of a wide range of conjugated polymer architectures such as homopolymers, donor-acceptors copolymers and porous polymers. However, the vast majority of direct arylation polymerization methodologies rely on the use of noble metals such as palladium (Pd), which is a costly, low abundant, relatively toxic, and unsustainable metal. To circumvent this, Thompson and co-workers were inspired by the small molecule copper catalyzed aryl-aryl cross-coupling for various iodinated arenes and electron-deficient heterocycles. To transition from small molecules to conjugated polymers, the group judiciously optimized the reaction conditions including the concentration, the nature of the solvent, the ligand, the temperature and the base employed. Conjugated polymers were then prepared in very high yields (up to 97%) with Mn of up to 10 kDa. NMR spectroscopy was used to characterize the recovered polymer products and confirmed the absence (or minimization) of undesired couplings. This report is the first example of perfectly alternating donor-acceptor conjugated polymers using copper catalyzed direct arylation polymerization thus offering an initial step towards the replacement of toxic metals like Pd. Future work will hope to address milder reaction conditions, lower catalyst loading, and a broader scope.
Tips/comments directly from the authors:
1. Fresh CuI is recommended. It is recommended to acquire a fresh bottle or a freshly purified stock of CuI and store it under inert gas in a freezer.
2. The strict exclusion of air and moisture is advisable, and so care should be taken to thoroughly sparge the reaction vessel with inert gas before and after the addition of reagents.
3. The selection of base is critical for the polymerization, and the base should be finely ground and dried using a vacuum oven before use. The base can be stored in a desiccator after drying, but should be dispensed quickly to limit contact with moisture
4. After addition of the CuI, the copper-phenanthroline catalyst appears to form instantly. However, stirring at room temperature for a several minutes before heating may help ensure complete coordination of the phenanthroline to copper
This article is FREE to read and download until 21st September 2018
Copper catalyzed synthesis of conjugated copolymers using direct arylation polymerization Polym. Chem., 2018, 9, 4120-4124, DOI: 10.1039/C8PY00913A
About the Web Writer
 Dr. Athina Anastasaki is a Web Writer for Polymer Chemistry. She is currently a Global Marie Curie Fellow working alongside Professor Craig Hawker at the University of California, Santa Barbara (UCSB). In January 2019, she will join the ETH Materials Department as an Assistant Professor to establish her independent group.
Dr. Athina Anastasaki is a Web Writer for Polymer Chemistry. She is currently a Global Marie Curie Fellow working alongside Professor Craig Hawker at the University of California, Santa Barbara (UCSB). In January 2019, she will join the ETH Materials Department as an Assistant Professor to establish her independent group.








")