Dou and co-workers report a new way to improve ultrasound responsiveness in polymeric self-assemblies.
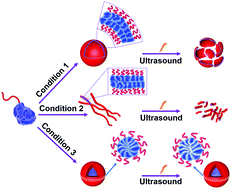
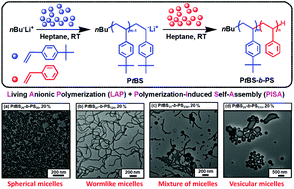
Polymer assemblies or nanoparticles hold great potential to improve diagnosis and treatment of diseases by encapsulating chemotherapeutic or imaging agents with masked toxicity and triggerring release at target sites. To release encapsulated agents, polymer assemblies are often composed of specific stimuli-responsive polymers that can change their properties upon response to external stimuli such as pH, temperature, light, redox, magnetic, and ultrasound. However, this approach limits the components of polymer nanoparticles to stimuli-responsive polymers. In this work, Chen and co-workers elegantly crosslink a common non-responsive diblock copolymer using an ultrasound-responsive crosslinker, followed by the preparation of polymer assemblies that can dissociate under gentle ultrasound treatment. In particular, the photodimerization of coumarin groups under UV irradiation (365 nm) triggered the crosslinking, and a subsequent ultrasound treatment (5 min treatment by the ultrasound of 20-25 kHz at 32.5 W) dissociated the resultant polymer nanoparticles. Interestingly, this strategy could be successfully applied to not only spherical micelles but also worms and vesicles. The use of ultrasound-responsive crosslinker reported in this work paves the way for synthesizing ultrasound-responsive polymer nanoparticles from any block copolymer (not limited to a few ultrasound-responsive copolymers), thus representing a major step forward in the synthesis of smart polymer nanoparticles for biological science and technology.
Read this article for FREE until 15th July!
Citation to the paper: A general method to greatly enhance ultrasound-responsiveness for common polymeric assemblies, Polym. Chem., 2020, 11, 3296-3304, DOI: 10.1039/d0py00254b
You can read the paper here.
About the web writer
 Dr. Athina Anastasaki is an Editorial Board Member and a Web Writer for Polymer Chemistry. Since January 2019, she joined the Materials Department of ETH Zurich as an Assistant Professor to establish her independent research group.
Dr. Athina Anastasaki is an Editorial Board Member and a Web Writer for Polymer Chemistry. Since January 2019, she joined the Materials Department of ETH Zurich as an Assistant Professor to establish her independent research group.








")