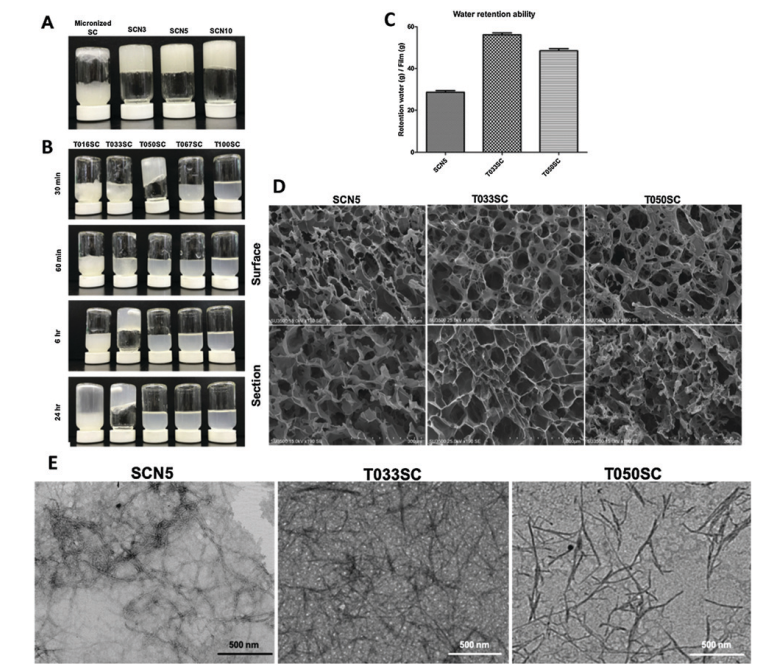
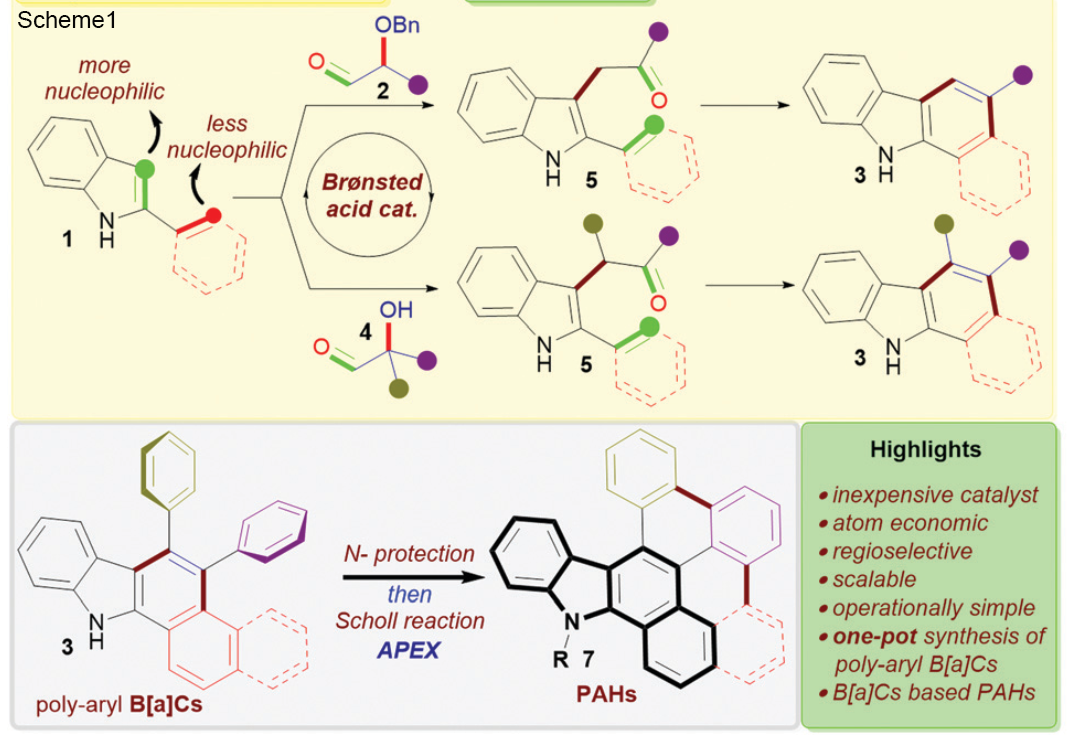
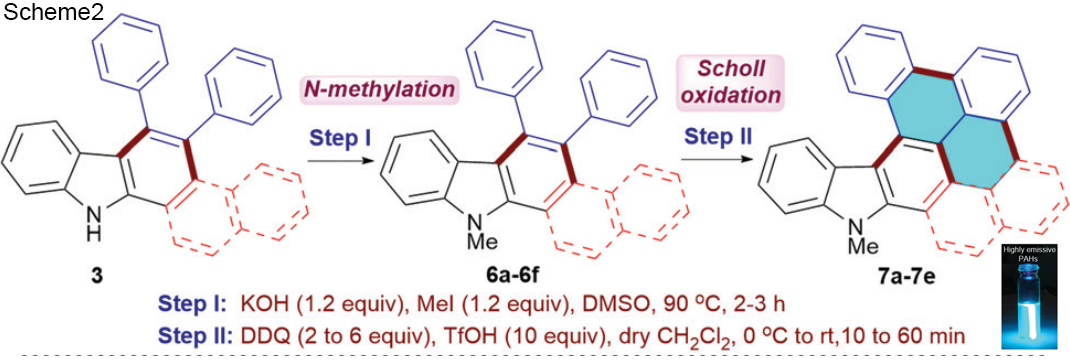
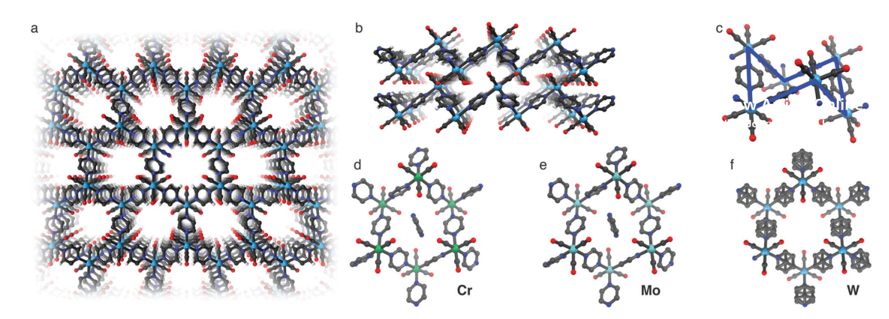
Antibodies emerge as a key player for disease detection and biologics due to its specificity of interaction and high binding affinity at target site. The application site for antibodies restricted to extracellular compartment as it has a poor plasma membrane permeability. Vehicles for direct intracellular delivery of antibodies are an extremely important alternative approach and various route have been explored including nanocarriers, liposomes, cell-penetrating peptides (CPPs). For most of these cases, loss of protein activity due to endolysosomal trapping or loss of protein activity lowers the efficacy of the antibody. So, it is highly desirable to formulate a fabrication strategy that is easily operable and antibody can be directly delivered to cytosol. In this aspect, cell-penetrating poly(disulfide)s (CPDs) which consist of disulfide polymer backbone with arginine-rich side chains permits thiol-mediated cellular uptake, which is insensitive to endocytosis inhibition, ensuing an efficient cytosolic availability. A team of researcher from Zhejiang University of China, reported a pH-responsive monomer to form new CPDs for enhanced intracellular delivery of antibodies into cancer cells. They tried to explore the stimuli responsiveness (lower pH) of tumor environment compared to healthy cells.

They have replaced the positively charged arginine moiety with neutral imidazole-based side chains. The resulting neutral CPD converts to positive charge upon protonation of the imidazole groups in acidic tumor environment. The advantage of charge reversal is easier cellular uptake by a combination of thiol-mediated and counter ion-activated uptake without significant endosomal trapping. The authors used a GSH-controlled NIR probe labelled at the N-terminal of the cargo protein so that the CPD could insert the cell followed by spectroscopic signal activation. Live-cell imaging of cancer cells using confocal laser scanning microscopy (CLSM) showed higher green fluorescence at pH 6.5 using IgGFITC-CPDIMD than neutral pH. This suggests higher uptake of imidazole based CPDs into cancer cells.
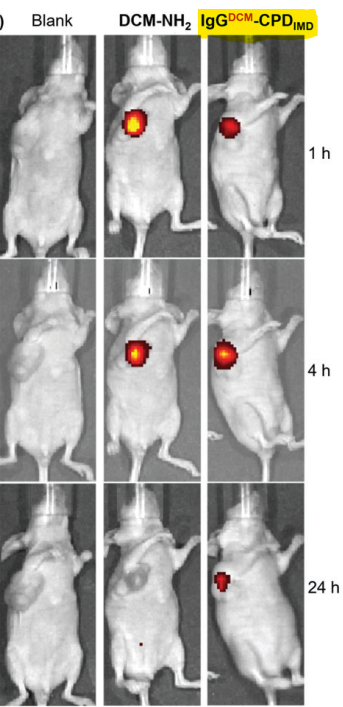
Fig2: In-vivo results show long-term effect by using the synthesized conjugate
For in-vivo experiments, the authors used GSH-activatable NIR fluorophore DCM-NH2 and attached it to CPDIMD. In vivo imaging of mice after intratumoral injection of the conjugate shows fluorescence signal at 1 h, that became stronger at 4 h, which indicate high drug dose accumulation at the tumor site. Both live-cell and in vivo results showed the great potential of this strategy for trackable and cancer-selective protein delivery with immediate cytosolic bioavailability. This new class of CPDs are expected to open an efficient platform for future cancer theranostics.
For details, please follow the article Chem. Commun., 2022, 58, 1314
About the blogger:

Dr Damayanti Bagchi is a postdoctoral researcher at University of California, Los Angeles, United States. She has obtained her PhD in Physical Chemistry from Satyendra Nath Bose National Centre for Basic Sciences, India. Her research is focused on spectroscopic studies of nano-biomaterials. She is interested in exploring light enabled therapeutics. She enjoys travelling and experimenting with various cuisines, which she found resembles with products/ side products of chemical reactions!
You can find her on Twitter at @DamayantiBagchi.








")