It turns out that rhenium-based compounds have been showing some promising anticancer activity as they’re stable, allow for real time imaging, structurally diverse, and have low off-site toxicity. The most commonly studied complexes are based around a Re(I) tricarbonyl core with the other three binding sites occupied by ligands of varying complexity. Researchers in the US and Australia developed a tricarbonyl Re isonitrile polypyridyl complex fac-[Re(CO)3(dmphen)(para-tolyl-isonitrile)]+, where dmphen = 2,9-dimethyl-1,10-phenanthro-line, called TRIP for short. TRIP showed promising cytotoxicity and can be imaged using confocal fluorescence microscopy, taking advantage of the emissive metal to ligand charge transfer (MLCT) state. The persistence of the emission indicates that the ligands remain bound to the Re even within cells. The complex’s cytotoxicity stems from its inducement of cells to accumulate misfolded proteins, resulting in apoptosis from the unfolded protein response (UPR). UPR induced cell death is relatively uncommon and led the researchers to find a method to characterize the speciation of TRIP in vitro. They used synchrotron X-ray fluorescence microscopy (XFM) to probe the cellular uptake and distribution of TRIP and an iodo-derivative I-TRIP by looking at elemental signals.

Figure 1. Chemical structures of TRIP and I-TRIP
I-TRIP is particularly well-suited to this type of study, as the iodine provides an additional spectroscopic handle on the isonitrile ligand absent in TRIP. Of course, the researchers had to confirm that I-TRIP possessed similar cytotoxicity and working mechanism to TRIP. Various assays and biological studies showed evidence of comparable cytotoxicity and mechanism, demonstrating that altering the substitution of the isonitrile ligand doesn’t significantly impact the bioactivity of the complex. With that settled, the experiments could move to the synchrotron to probe elemental distributions.
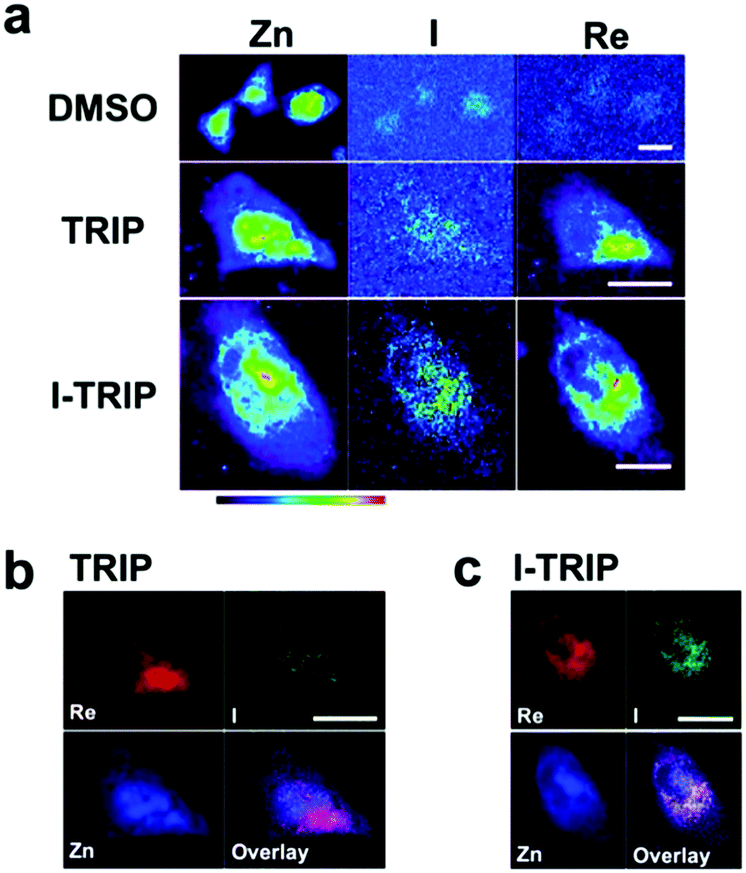
Figure 2. XFM elemental distribution maps of HeLa cervical cancer cells treated with either DMSO (control), TRIP, or I-TRIP.
Cells treated with both TRIP and I-TRIP show a clear Re signal, confirming that they can enter and persist in cells. Critically, the colocalization of the Re and I maps for I-TRIP samples indicate that the isonitrile ligand remains bound as a part of the Re complex inside the cells. This strongly suggests that the Re complex is intact while it induces cell death, adding to the developing mechanistic understanding of their activity. This work shows the utility of XRM as a technique to study the distribution of organometallic complexes in living cells. Additionally, the tunability and stable bioactivity of the Re complexes shows that they’re amenable to study by a wide range of techniques that will allow for further mechanistic probing.
To find out more, please read:
Chilaluck C. Konkankit, James Lovett, Hugh H. Harris and Justin J. Wilson
Chem. Commun., 2020, 56, 6515-6518
About the blogger:
 Dr. Beth Mundy is a recent PhD in chemistry from the Cossairt lab at the University of Washington in Seattle, Washington. Her research focused on developing new and better ways to synthesize nanomaterials for energy applications. She is often spotted knitting in seminars or with her nose in a good book. You can find her on Twitter at @BethMundySci.
Dr. Beth Mundy is a recent PhD in chemistry from the Cossairt lab at the University of Washington in Seattle, Washington. Her research focused on developing new and better ways to synthesize nanomaterials for energy applications. She is often spotted knitting in seminars or with her nose in a good book. You can find her on Twitter at @BethMundySci.








")