I don’t know if you’ve looked at the structure of pharmaceuticals recently, but most novel drugs are rather complicated. Identifying promising new targets is just the start for synthetic chemists; they then need to figure out how to use a series of reactions to take simple (and commercially available) molecules and transform them into a new drug. They also must predict all possible side reactions and products given a set of reaction conditions, particularly when a range of functional groups are involved. Historic approaches involved manual curation of reaction rules, limited by personal experience and the state of the accessed chemical literature. Newer approaches seek to create templates directly from data but are defined by available data sets and cannot reliably extrapolate. The emergence of machine learning offers the opportunity to move beyond traditional templating and atom mapping of reactants to products. It also offers to take full advantage of novel technologies and address problems with dataset bias and ineffective modeling systems.
In a collaboration between academics in the UK and industrial scientists in the US, researchers used Molecular Transformer, an attention-based machine translation model, to perform both reaction prediction and retrosynthesis analysis after training on a publicly available dataset. Instead of atom mapping, which moves atoms from the reactants to the products, Molecular Transformer (MT) relies on SMILES text strings, which represent structures in a line format. A unique aspect of this work is the validation and training performed using proprietary data of drug targets from Pfizer. They used three datasets: the first a literature standard from the US Patent and Trade Office (USPTO), the second from internal medicinal chemistry projects in Pfizer, and the final a diverse range of 50,000 reactions from US patents (USPTO-R). Building on previous research from the authors, they trained the MT on both the Pfizer data and the initial USPTO data sets. They found that the Pfizer data provided the most accurate product predictions and that the MT could also return a confidence rating to determine the probability the prediction is correct.
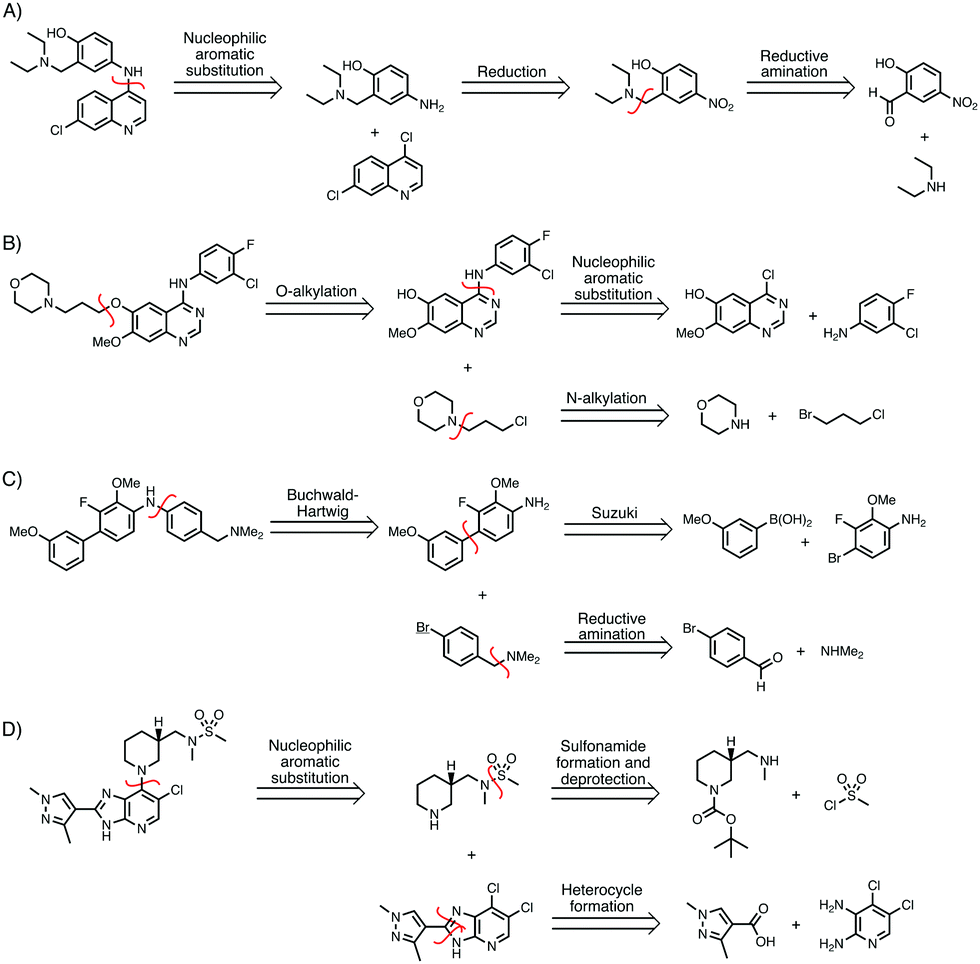
Figure 1. Sample syntheses predicted by Molecular Transformer for various bioactive molecules of interest.
While synthesis predictions can easily be checked, it’s harder to confirm accuracy with retrosynthesis since there is not a single correct answer. The researchers used the broad USPTO-R to train MT, which consistently outperformed both a benchmark template-based program and another literature machine learning method also trained on USPTO-R. When tested on the Pfizer dataset, the MT performed best with 31.5% accuracy despite the datasets coming from different regions of chemical space (which increased to 91% when MT was trained on Pfizer data). Figure 1 shows several predicted routes for the synthesis of bioactive molecules as predicted by MT, which generally agree with established syntheses. These data suggest the highly generalizable nature of MT as a tool for developing novel pharmaceutically interesting molecules.
To find out more, please read:
Molecular Transformer unifies reaction prediction and retrosynthesis across pharma chemical space
Alpha A. Lee, Qingyi Yang, Vishnu Sresht, Peter Bolgar, Xinjun Hou, Jacquelyn L. Klug-McLeod and Christopher R. Butler
Chem. Commun., 2019, 55, 12152-12155.
About the blogger:
 Beth Mundy is a PhD candidate in chemistry in the Cossairt lab at the University of Washington in Seattle, Washington. Her research focuses on developing new and better ways to synthesize nanomaterials for energy applications. She is often spotted knitting in seminars or with her nose in a good book. You can find her on Twitter at @BethMundySci.
Beth Mundy is a PhD candidate in chemistry in the Cossairt lab at the University of Washington in Seattle, Washington. Her research focuses on developing new and better ways to synthesize nanomaterials for energy applications. She is often spotted knitting in seminars or with her nose in a good book. You can find her on Twitter at @BethMundySci.








")