Making catalysts from non-precious metals has been a challenge embraced by chemists over the past few decades. In particular, the ability of alkaline earth metal catalysts to be both highly stable and highly reactive has made them attractive for study. Magnesium complexes can catalyze hydroelementation across a carbon-oxygen double bond with a wide scope of substrates. However, it isn’t enough to just perform this reaction. For meaningful reactivity the catalyst would be enantioselective and prior to this report only one example of a stereoselective magnesium-based system existed in the literature. A persistent barrier to developing this reactivity has been the propensity of alkaline earth metal hydrides (a likely step in the catalytic cycle) to form contact-ion pairs with available anions. Based on this framework, researchers in Germany developed a catalyst motif involving borohydride-alkaline earth metal adduct formation to enhance stereoselectivity.
They drew on their precious work utilizing manganese complexes that demonstrated enantioselectivity, applying it to the magnesium system. They could straightforwardly create the magnesium alkyl complexes by mixing ligand and magnesium alkyl precursors. The complexes were tested for hydroelemetation using acetone as a model substrate. The rate and selectivity of the reaction was significantly impacted by the choice of reducing agent, with non-boron reductants producing low yields and low enantioselectivity. Altering the ligand backbone didn’t improve selectivity, but purifying an assembled precatalyst and slowly raising the temperature from -40 oC to RT increased enantiomeric excess to 96%. Even more exciting, using this general reaction format high enantioselectivity was seen for a wide range of aryl alkyl ketones screened with no adverse reactivity seen towards ester moieties (Figure 1). This system also catalyzes reactions with α-substituted, cyclic alkyl aryl, and dialkyl ketones, but with lowered enantioselectivity. However, the catalyst is tolerant of a range of functional groups and remains selective for carbonyls implying broad possible utility.
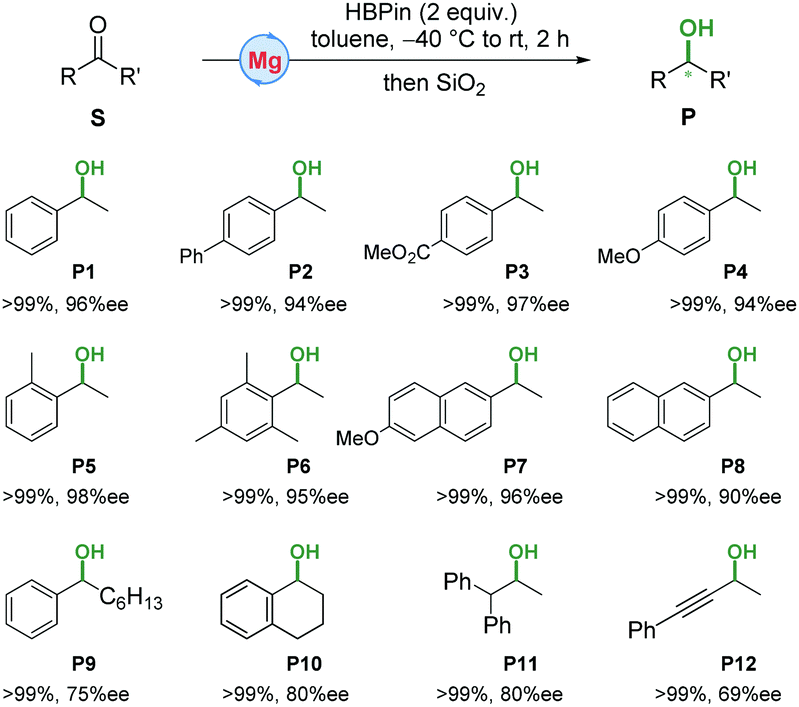
Figure 1. Substrates tested for hydroelementation using a magnesium catalyst with conversion and stereoselectivity numbers.
To more fully understand the system, the researchers undertook experiments to find possible intermediates in the catalytic cycle. The reactivity at room temperature required consistent low temperature work to prevent further progress of the intermediates along the catalytic cycle. In the absence of a boron-derived reducing agent, the catalyst forms a highly labile, and expected, alkoxide intermediate that is not observable under catalytic conditions. When the precatalyst is reacted with excess borane it forms a borohydride intermediate that was crystallographically observed.

Figure 2. Solid state molecular crystal structure of borohydride intermediate.
When this complex was reacted with a fluorinated ketone, variable temperature boron and fluorine NMR was used to identify transient species. Upon addition of the ketone immediate shifts in the boron signal to two species, one of which remains when the temperature increases to -60 oC. The unstable species can be attributed to a ketone borohydride complex, but the dynamic nature of the system makes crystallizing these intermediates very challenging. DFT calculations suggest a low-energy transition state that favors the (S)-product and accounts for the catalyst’s consistent stereoselectivity.
To find out more, please read:
Borohydride intermediates pave the way for magnesium-catalysed enantioselective ketone reduction
Vladislav Vasilenko, Clemens K. Blasius, Hubert Wadepohl and Lutz H. Gade
Chem. Commun., 2020, 56, 1203-1206
About the blogger:
 Beth Mundy is a PhD candidate in chemistry in the Cossairt lab at the University of Washington in Seattle, Washington. Her research focuses on developing new and better ways to synthesize nanomaterials for energy applications. She is often spotted knitting in seminars or with her nose in a good book. You can find her on Twitter at @BethMundySci.
Beth Mundy is a PhD candidate in chemistry in the Cossairt lab at the University of Washington in Seattle, Washington. Her research focuses on developing new and better ways to synthesize nanomaterials for energy applications. She is often spotted knitting in seminars or with her nose in a good book. You can find her on Twitter at @BethMundySci.








")