Zeolites, a class of porous alumina-silicate materials, are industrially critical adsorbents and catalysts. Their highly robust nature and wide range of structural types (over 200!) make them suited to a range of applications. In particular, the general zeolite topology and pore size are selected to match and stabilize the intermediates of a chemical reaction. However, the tunability of zeolites is limited when compared to molecular catalysts, making them more like a solvent than, say, an enzyme. An active field of research is bridging the gap between the robust, scalable zeolites and highly controllable homogenous catalysts. Recent work identified organic residues maintained with the zeolite pores as key in the transformation of methanol to hydrocarbons. Previous fundamental studies demonstrated that a wide range of carbonyl and carbonyl derivative compounds promote the dehydration of methanol to dimethyl ether (DME).
Researchers at BP used methyl mono- and di-carboxylate esters to dehydrate methanol to DME at low temperatures. The mild reaction conditions allowed for high selectivity for DME while eliminating convoluting side reactions. They added either methyl formate or methyl n-hexanoate to a series of zeolite with pores ranging from narrow to wide. At a 5 mol% concentration relative to methanol they saw significant increases in DME production, particularly for the medium and wide pores. Systematic testing of carboxylate chain length found that increasing chain length increased turnovers occurred until methyl n-hexanoate, after which no further benefits were observed as the n-methyl hexanoate had already saturated the catalyst (Figure 1). All proved highly selective for converting methanol to DME with no observed hydrocarbon formation.
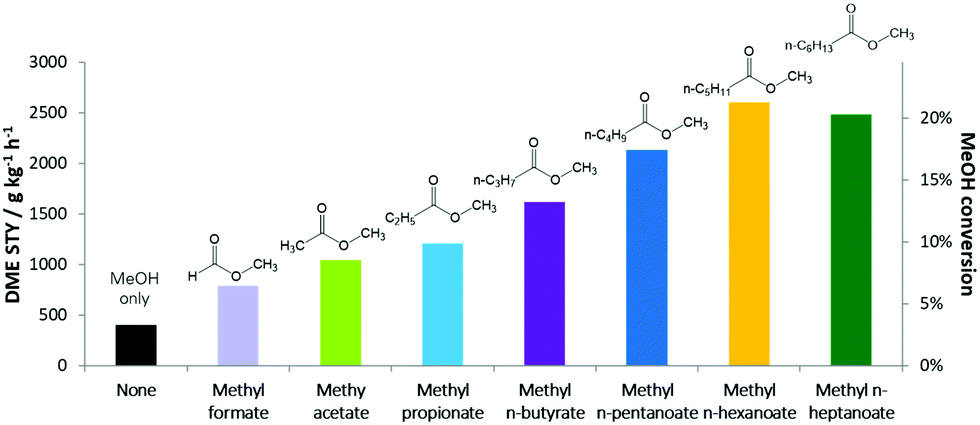
Figure 1. Production of DME on a medium-pore zeolite with methyl carboxylate esters of varying chain lengths.
The experimental results were coupled with theoretical work modeling the energetics of the adsorption of the ester onto the zeolite. The calculations showed an increase in adsorption energy with increased chain length, attributed to van der Waals interactions.
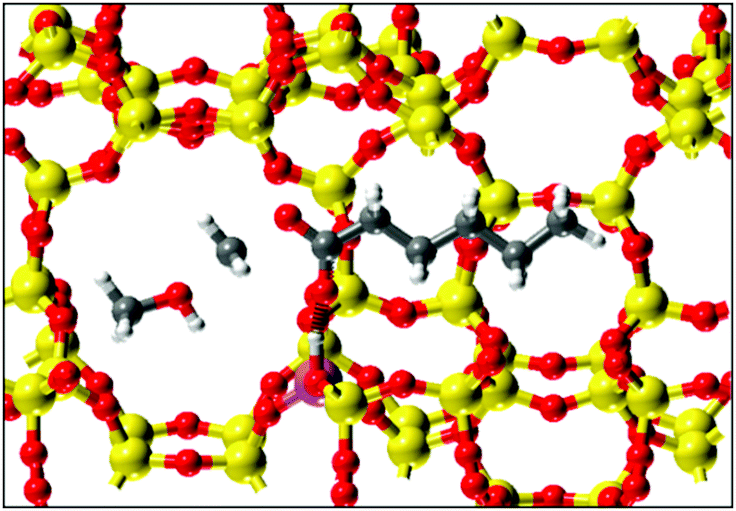
Figure 2. Transition state predicted by molecular modeling with methanol attacking the organic promoter adsorbed on the zeolite catalyst.
They also gave even higher energies to molecules with two carboxylate esters, like dimethyl adipate. In fact, the strongly binding molecules produced increased catalysis at loadings as low as 0.001% with respect to methanol. The promoters can be easily switched by changing the input, demonstrating the reversibility of binding at the active site. Additional molecular modeling was used to study possible transition states to develop a catalytic cycle. A proposed transition state involves a direct reaction between the methanol and the organic promotor, however specific evidence has yet to be seen. Additional work examining the role of the water present as a co-adsorbate and its impacts on transition states has yet to be done. Overall, the use of various organic molecules as promotors for the dehydration of methanol to DME on various zeolite catalysts was explored. This represents exciting fundamental study of industrially-relevant chemistry with significant room for future work.
To find out more, please read:
Benjamin J. Dennis-Smither, Zhiqiang Yang, Corneliu Buda, Xuebin Liu, Neil Sainty, Xingzhi Tan and Glenn J. Sunley
Chem. Commun., 2019, 55, 13804-13807.
About the blogger:
 Beth Mundy is a PhD candidate in chemistry in the Cossairt lab at the University of Washington in Seattle, Washington. Her research focuses on developing new and better ways to synthesize nanomaterials for energy applications. She is often spotted knitting in seminars or with her nose in a good book. You can find her on Twitter at @BethMundySci.
Beth Mundy is a PhD candidate in chemistry in the Cossairt lab at the University of Washington in Seattle, Washington. Her research focuses on developing new and better ways to synthesize nanomaterials for energy applications. She is often spotted knitting in seminars or with her nose in a good book. You can find her on Twitter at @BethMundySci.








")