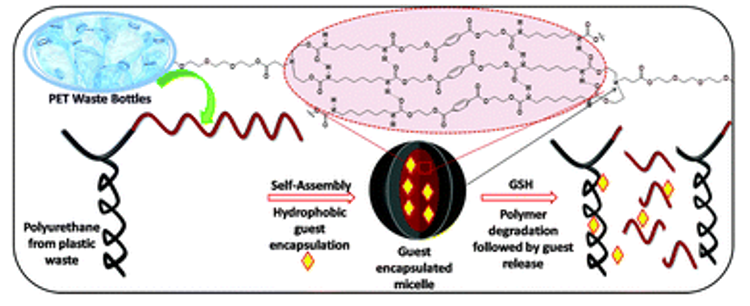
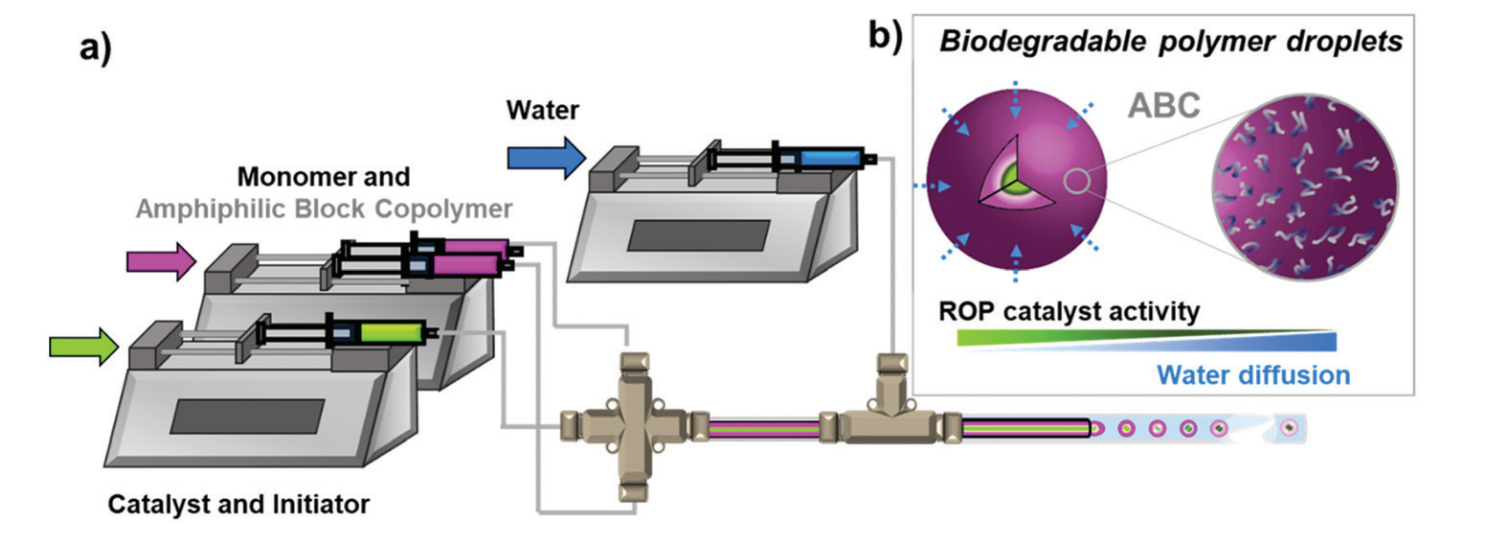
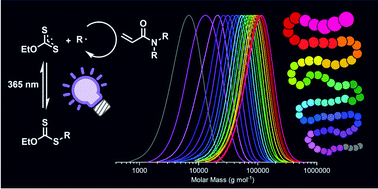
Abad et al. develop formulations of self-assemblies containing nucleobase analogues via seeded RAFT in water.
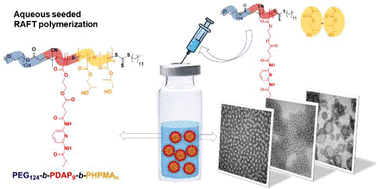
Polymer chemists have long exploited the specific hydrogen bonding interactions between nucleobase pairs to control polymer structure or sequence, to template polymerizations or drive self-assembly. Although several approaches have been employed for the synthesis of nucleobase containing polymers, the poor solubility of nucleobase-containing monomers has hampered their polymerization in water.
To address this, Blasco, Piñol and collaborators synthesized a diblock copolymer containing poly(ethylene glycol) (PEG) and 2,6-diacylaminopyridine (DAP) polymethacrylate via RAFT. Upon dispersing in water this macro-CTA agent was used for the aqueous seeded RAFT polymerization of 2-hydroxypropyl methacrylate (HPMA). Furthermore, a phase diagram that correlates the degree of polymerization and solid concentration with the morphologies of the resulting self-assemblies was constructed. Through this systematic study, low to high order morphologies (from spherical micelles to worms and to vesicles) could be observed. Interestingly all morphologies proved to be stable for extended periods of time with the exception of worms found to turn into spherical micelles after few weeks. To exploit the ability to functionalize the DAP moieties through H-bonding during aqueous seeded RAFT polymerization, a cross-linker bearing four thymine terminal groups was used. Finally, the higher stability of the assemblies produced via supramolecular cross-linking was studied via encapsulation and subsequent release of the hydrophobic probe Nile Red.
In summary, this study provides a metal-free methodology to produce self-assemblies containing nucleobase analogues in high concentrations via aqueous seeded RAFT polymerization. The ability to control assembly, functionalize via exploiting supramolecular interactions and load with cargo, enhances their potential use as nanocarriers.
Tips/comments directly from the authors:
- This new strategy integrating non-water soluble groups, such as DAP units, into a BC enabled the preparation of highly concentrated aqueous self-assembly dispersions using the PISA methodology.
- The DAP units were further exploited for supramolecular H-bonding functionalization with cross-linker containing complementary thymine groups.
- Previous work on amphiphilic block copolymers having DAP units has proved their potential to prepare stimuli-responsive self-assemblies of interest in nanomedicine by nanoprecipitation or microfluidic. This article takes an important step forward since the potential of the polymers is upgraded with the processing of highly concentrated dispersions by this new straightforward strategy.
- This paper is the result of a collaborative effort between the groups at University of Zaragoza (Spain) and Heidelberg University (Germany)
Citation of the paper: Aqueous seeded RAFT polymerization for the preparation of self-assemblies containing nucleobase analogues, Polym. Chem., 2023,14, 71-80.
Link to the paper: https://pubs.rsc.org/en/content/articlelanding/2023/py/d2py01250b
Link to authors website (or social media)
https://liquidcrystals.unizar.es/ @clip_group_lab (Twitter)
https://www.imseam.uni-heidelberg.de/blasco @EvaBlascoPo (Twitter)
 |
Dr. Kelly Velonia is an Advisory Board Member and a Web Writer for Polymer Chemistry. She joined the Department of Materials Science and Technology at the University of Crete in 2007. Research in her group focuses on the synthesis and applications of bioconjugates and biopolymers. You can follow Kelly on twitter @KellyVelonia.
|








")