Mertens et al. synthesized a series of sequence-defined oligoampholytes and evaluated their properties.
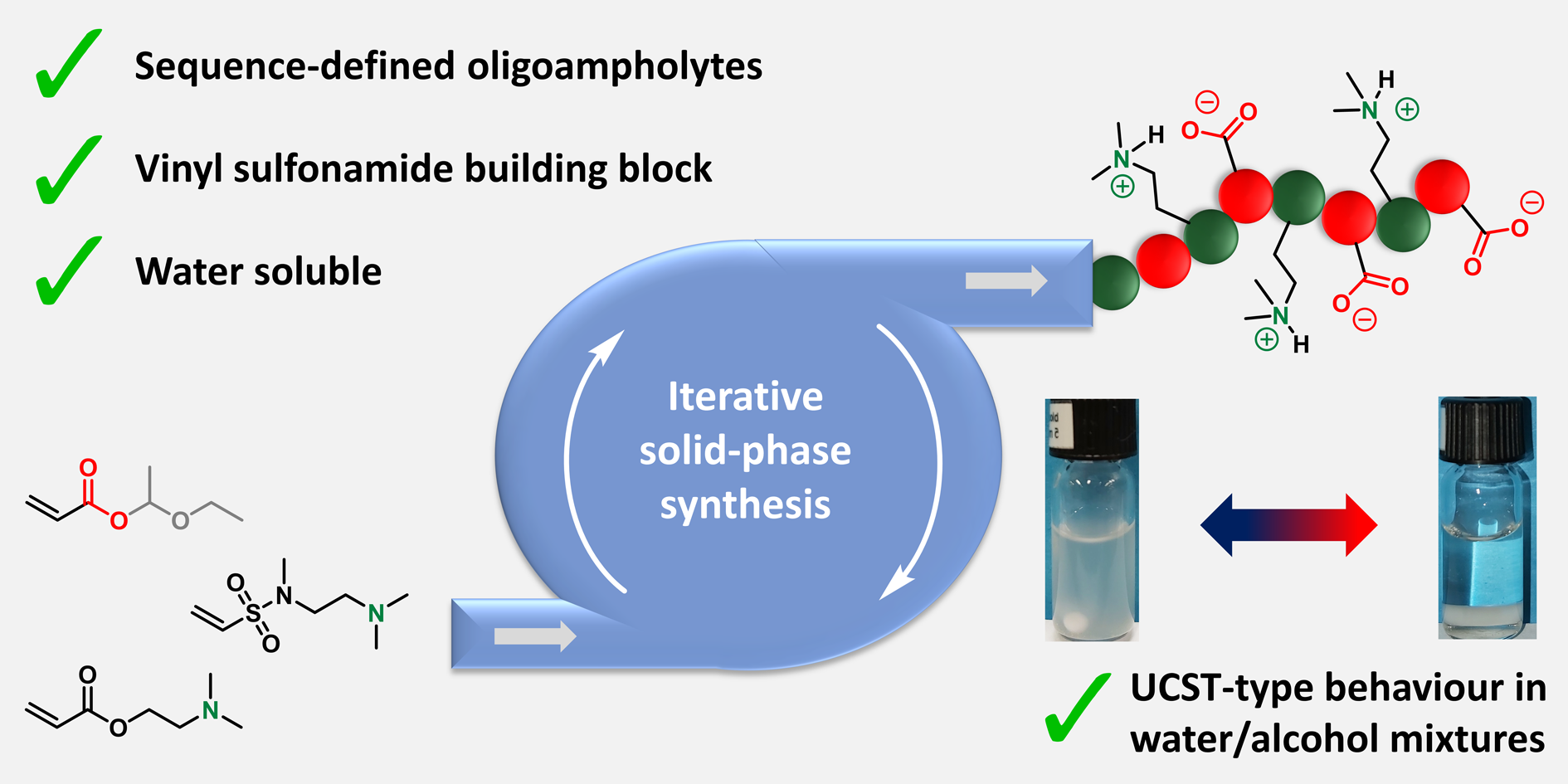
Polyampholytes are polymers compromising of positively and negatively charged groups and present a number of distinct properties which allows them to be employed in many applications including antifouling coatings and drug delivery systems. Polyampholytes are usually produced by conventional polymerizations strategies yielding materials with varied chain lengths and monomer composition, thus compromising the purity and the properties of these molecules. Du Prez’s group has contributed to this field by developing a chemical platform based on thiolactone chemistry which allowed the introduction of a variety of side-chain functionalities employing acrylic, amine or halide building blocks. In their current contribution, novel sequence-defined oligoampholytes with carboxylic acid and tertiary amine side-chains were elegantly synthesized through a thiolactone-based solid-phase synthesis protocol, in which the side-chain functionalities were incorporated through a thiol-Michael reaction. Although initially acrylate-based building blocks were employed to introduce the tertiary amine, severe side reactions were observed including hydrolysis or methanolysis. Instead, a tertiary amine bearing vinyl sulfonamide proved to be an efficient and stable alternative for the used acrylates and thus could be directly integrated into the synthetic protocol. A wide range of oligomers (up to a hexamer) were synthesized with an alternating or block-like arrangement of the tertiary amine and acid side-chains. These perfectly uniform oligomers were shown to be soluble in water and exhibited UCST-type thermoresponsive behaviour in alcoholic/aqueous mixtures. Such properties were influenced by both the length and the sequence of the oligoampholytes. As the authors elude in their conclusion, the ability to fully control the arrangement of the amphiphilic groups is a powerful tool to design the properties and functions of these uniform macromolecules.
Tips/comments directly from the authors:
- The tertiary amine side-chains are attached to the backbone through a β-thioester bond resulting from the thiol-Michael addition, which was found to be susceptible towards hydrolysis and methanolysis. Prolonging the carbon spacer between the ester and the tertiary amine reduced the rate of this undesired degradation, yet it could not be completely inhibited.
- Compared to acrylates, vinyl sulfonamides are a class of Michael acceptors that received considerably less attention within the area of polymer chemistry, although they can provide favourable properties. The thiol-vinyl sulfonamide adduct is stable towards hydrolysis, unlike an acrylate, while the reaction kinetics are comparable. These aspects are highlighted within the present work, as well as in the work of others.
- While we demonstrate the synthesis of these oligomers together with the fundamental characterisation of their properties, further research is required to establish in-depth structure-property relationships.
Citation to the paper: Sequence-defined oligoampholytes using hydrolytically stable vinyl sulfonamides: design and UCST behaviour, Polym. Chem., 2021,12, 4193-4204, DOI: 10.1039/D1PY00662B
Link to the paper:
https://pubs.rsc.org/en/content/articlelanding/2021/py/d1py00662b
 Dr. Athina Anastasaki is an Editorial Board Member and a Web Writer for Polymer Chemistry. Since January 2019, she joined the Materials Department of ETH Zurich as an Assistant Professor to establish her independent research group.
Dr. Athina Anastasaki is an Editorial Board Member and a Web Writer for Polymer Chemistry. Since January 2019, she joined the Materials Department of ETH Zurich as an Assistant Professor to establish her independent research group.








")