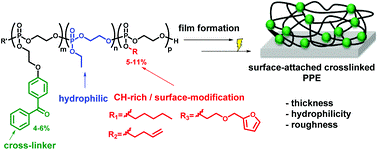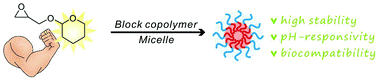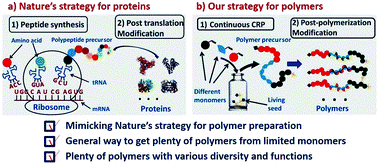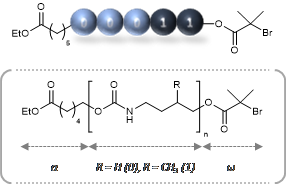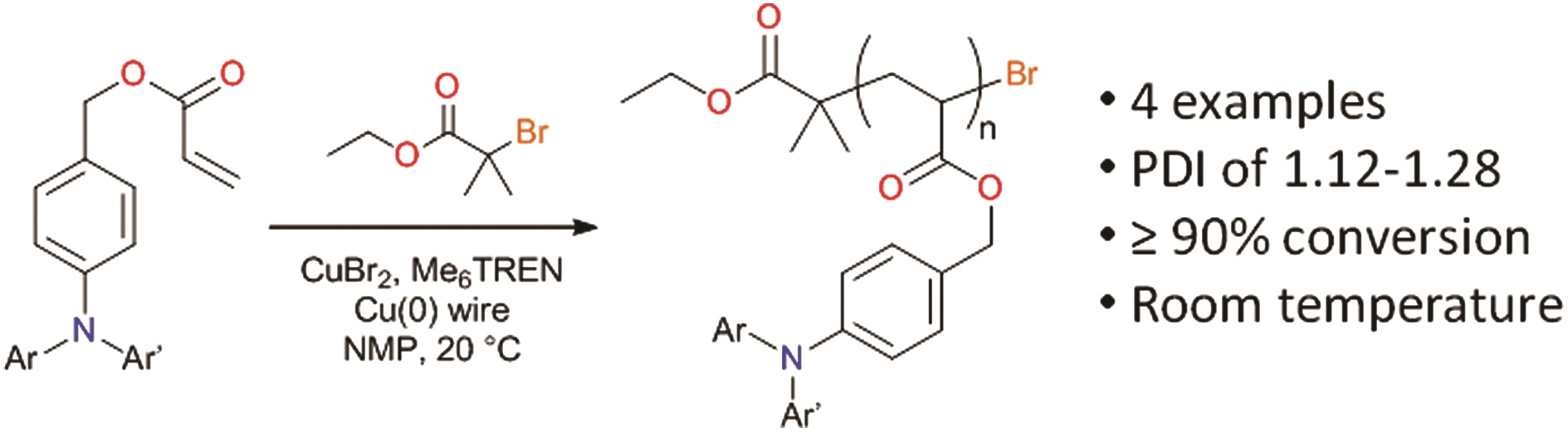
p-type organic semiconductor polymers can find a use in organic electronics, including organic light-emitting diodes (OLEDs), solar cells, and organic thin-film transistors. These materials offer unique characteristics over inorganic semiconductors such as flexibility and light weight. To maximize their potential, reversible deactivation radical polymerization (RDRP) methodologies are often used with traditional atom transfer radical polymerization and reversible addition/fragmentation chain transfer polymerization dominating in this area. To this end, Hudson and co-workers exploited Cu(0)-RDRP as an effective method for preparing functional acrylate-based polymers with p-type organic semiconductors as side chains. Impressively, all polymers were obtained in high yields (~ 90 %) with low dispersity and high end group functionality while the polymerizations displayed first order kinetics. Both low and high molecular weight polymers could be prepared in a facile manner and the choice of solvent seemed to be crucial to maintain good control over the molecular weight distributions. It should be highlighted that the described technique represents the most simple, low-cost and efficient way to synthesize these materials with improved end group functionality and yields over previous methods. The optical, electrochemical and thermal properties of each of these p-type materials were also carefully investigated with cyclic voltammetry and thermogravimetric analysis revealing the potential for further studies in optoelectronic applications. The Hudson group will now focus on the synthesis of more complex materials, including multiblock copolymers, and subsequently utilize them for optoelectronics.
Tips/comments directly from the authors:
- The Cu(0) wire should be prepared immediately before use for best activity, as substantial reductions in polymerization rate are observed when the wire is cleaned and stored.
- Reducing the relative amount of Cu(0) wire when attempting the synthesis of high molecular weight polymers reduces the polymerization rate, but provides improved control over the polydispersity of the products.
- For long-term storage all monomers should be stored in the freezer (–10 ºC), but are stable on the bench top under air for 1-2 days.
- Yields of pure monomers 5a-c are substantially improved when purification is conducted quickly (<5 min) on a short silica column to minimize decomposition; the same urgency is not required for 5d.
Cu(0)-RDRP of acrylates based on p-type organic semiconductors, Polym. Chem., 2018, 9, 1397-1403, DOI: 10.1039/C8PY00295A
This article is free to read until 30 April
About the webwriter
 Dr. Athina Anastasaki is a Web Writer for Polymer Chemistry. She is currently a Global Marie Curie Fellow working alongside Professor Craig Hawker at the University of California, Santa Barbara (UCSB). Please, visit this site for more information.
Dr. Athina Anastasaki is a Web Writer for Polymer Chemistry. She is currently a Global Marie Curie Fellow working alongside Professor Craig Hawker at the University of California, Santa Barbara (UCSB). Please, visit this site for more information.








")