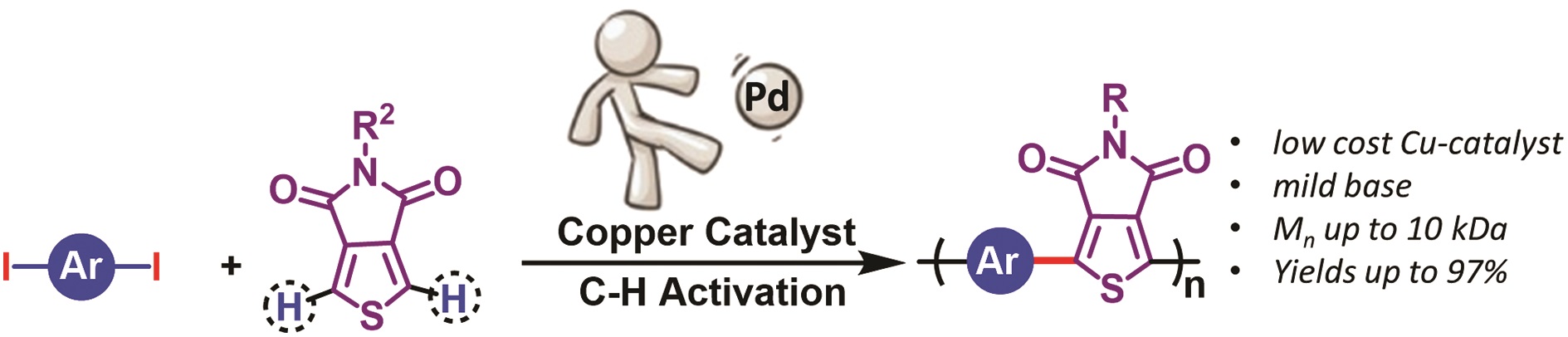
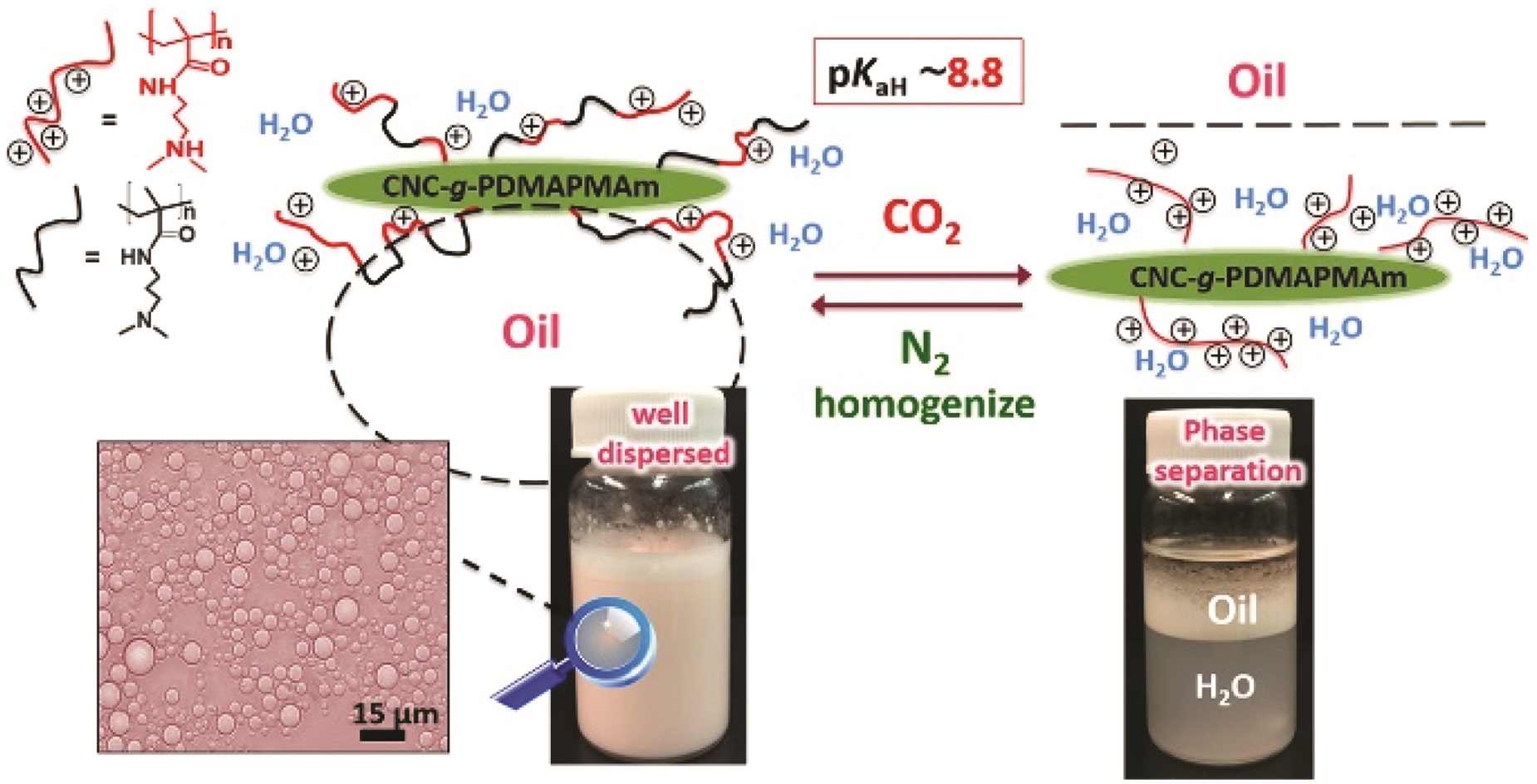
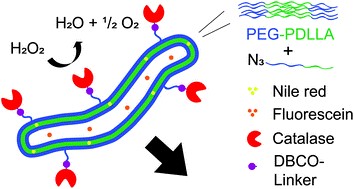
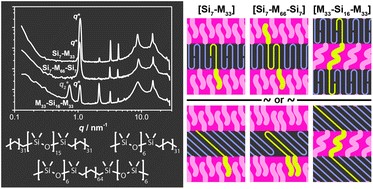
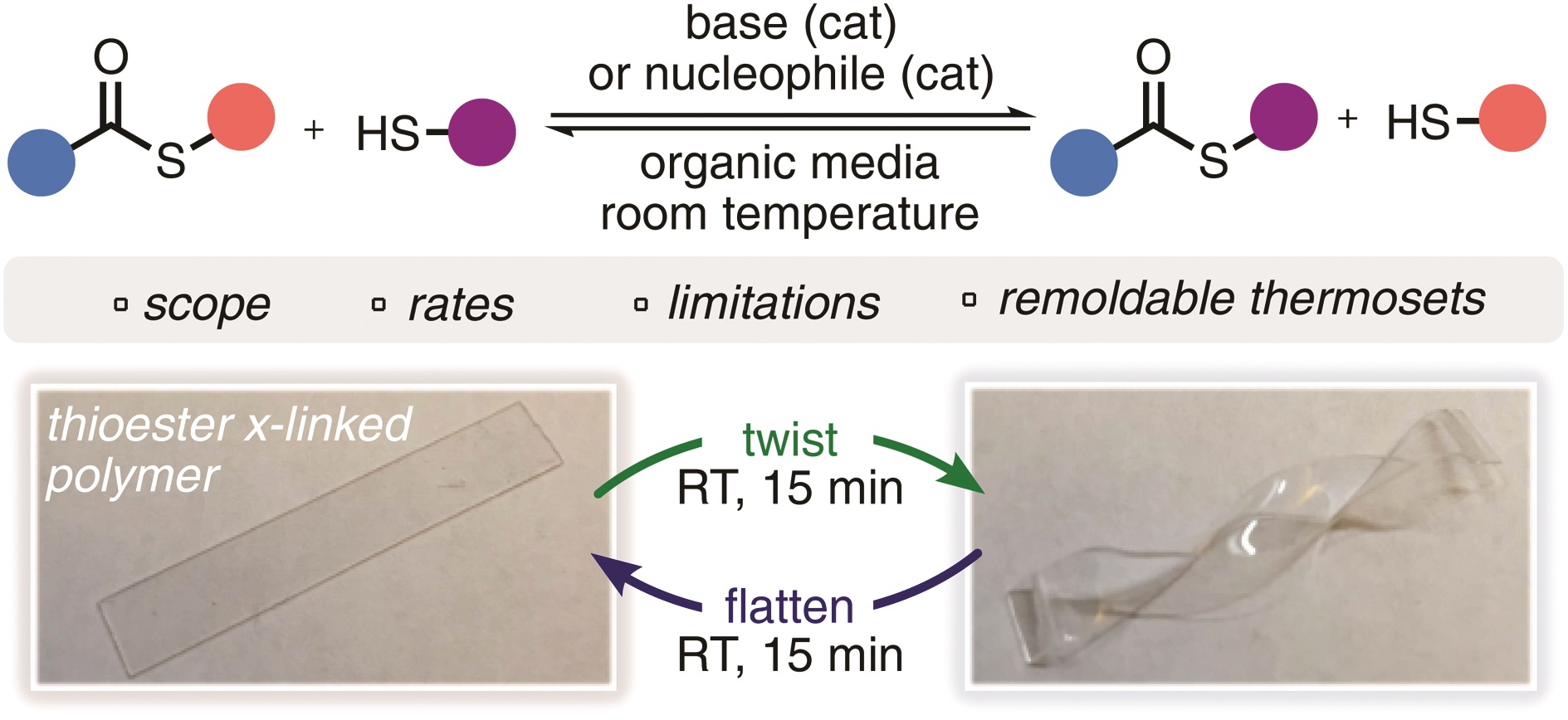
The thiol-thioester exchange is a common reaction in dynamic covalent chemistry. This reaction has been extensively optimized in aqueous media facilitating the widespread use in biochemistry and other bio-related applications. However, the utility of this reaction in material and polymer science is currently underexplored. This is possibly due to the fact that most polymer/material systems require organic media for their respective synthesis. To overcome this barrier and extend the scope and applications of thiol-thioester exchange, Bowman and co-workers explored this dynamic exchange in both small molecule and polymer analogues in a wide range of organic solvents. The effect of the pKa of the thiol and base employed, the electronic character of the thioester, the polarity of the solvent, the effect of the temperature and the nucleophilicity of the catalyst were thoroughly investigated. By judiciously choosing and optimizing all these parameters, the authors were able to tune the thiol-thioester exchange in both small molecules and subsequently network polymers to reduce applied stresses or change shape of the materials following polymerization. All these findings were thoroughly reported and explained by crafting an impressive “user’s guide” that can be useful for a large number of practitioners within the polymer/material community. The authors anticipate that the extremely robust, tuneable and responsive exchange reaction will further enable polymer/material scientists to develop new smart materials and will pave the way for further applications.
Tips/comments directly from the authors:
1. When comparing a panel of various thioesters exchanging with various thiols, the authors have found that thermodynamically the acyl group of a thioester prefers to rest on thiols of the highest pKa and will exchange rapidly to achieve this minima.
2. Base catalysts of a higher pKa form more thiolate and therefore promote the thiol-thioester exchange more rapidly in both small molecule systems and in crosslinked polymers, all things held the same. Base catalysts, if employed in larger concentrations, were, however, found to significantly retard the free radical thiol-ene reaction to produce crosslinked polymers. This can be overcome by optimizing the concentration of radical initiator (-photo, -thermal, or –redox) with respect to base.
3. Nucleophilic catalysts of a higher N-value (see Herbert Mayr’s work for more details on the calculation of this parameter) were found to promote the thiol-thioester exchange more rapidly in both small molecule systems and in crosslinked polymers, all things held the same. Unlike basic catalysts, nucleophilic catalysts did not show similar retardation in the free radical thiol-ene reaction to produce crosslinked polymers.
4. Due to polar intermediates formed during the thiol-thioester exchange reaction, polar solvents/matrices most effectively promote this dynamic exchange, especially with weaker bases.
5. Due to the low energetic barrier for the thiol-thioester exchange reaction, temperature does not have a great affect on the outcome of the exchange, however, it does improve the kinetics.
6. If the readers have any unanswered questions regarding this reaction, schemes for placing it into a polymer matrix, or other general queries, please direct them to brady.worrell@gmail.com and we’ll get to the bottom of it.
Read the full article for FREE until 30th October
A user’s guide to the thiol-thioester exchange in organic media: scope, limitations, and applications in material science, Polym. Chem., 2018, 9, 4523-4534, DOI: 10.1039/C8PY01031E
About the web writer
 Dr. Athina Anastasaki is a Web Writer for Polymer Chemistry. She is currently a Global Marie Curie Fellow working alongside Professor Craig Hawker at the University of California, Santa Barbara (UCSB). In January 2019, she will join the ETH Materials Department as an Assistant Professor to establish her independent group.
Dr. Athina Anastasaki is a Web Writer for Polymer Chemistry. She is currently a Global Marie Curie Fellow working alongside Professor Craig Hawker at the University of California, Santa Barbara (UCSB). In January 2019, she will join the ETH Materials Department as an Assistant Professor to establish her independent group.








")