A group of researchers at the Chinese Academy of Sciences and Southwest University want us to kick the fossil fuels habit. Their research comes to us from China, a country using roughly one quarter of the world’s yearly energy consumption, and where the finite nature of fossil fuels is a very real threat to energy supply security. Leading in energy use, China also leads the world in electricity production from renewable sources and investment in clean energy projects.
Hydrogen is considered a viable alternative to fossil fuels as it is energy rich, more so than petrol or ethanol at 39 kWh/kg (petrol: 13 kWh/kg, ethanol: 8.2 kWh/kg), and upon combustion emits only water vapour. However, hydrogen is often obtained from fossil fuels, and it will only be a practical option for the world’s future energy needs if it can be produced from a renewable source.
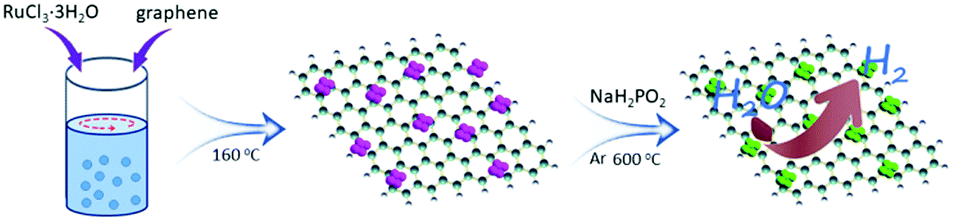
Preparation of the Ru2P/reduced graphene oxide catalyst
To this end, water splitting offers a solution. In a water electrolysis cell, hydrogen is produced at the cathode via the hydrogen evolution reaction (HER, 2H+ + 2e– –> H2), and molecular oxygen is produced at the anode (2H2O –> O2 + 4H+ + 4e–). It is ideal in theory, but high energy efficiencies are required to make water splitting viable, and this relies on the development of catalytic electrodes to minimize overpotentials required to drive the reaction. Currently, state of the art HER electrocatalysts use platinum, which is expensive and rare. Furthermore, platinum catalysts are most efficient in an acidic electrolyte and proceed 2-3 times slower in alkaline solutions. On the other hand, the best oxygen evolution catalysts perform better in alkaline environments. Using an alkaline electrolyte has overall advantages as it is less corrosive, thus increasing the stability and lifetime of the electrolytic cell.
The authors have developed a HER catalyst, using ruthenium, with overpotentials and current densities superior to Pt/C in both alkaline and acidic conditions.
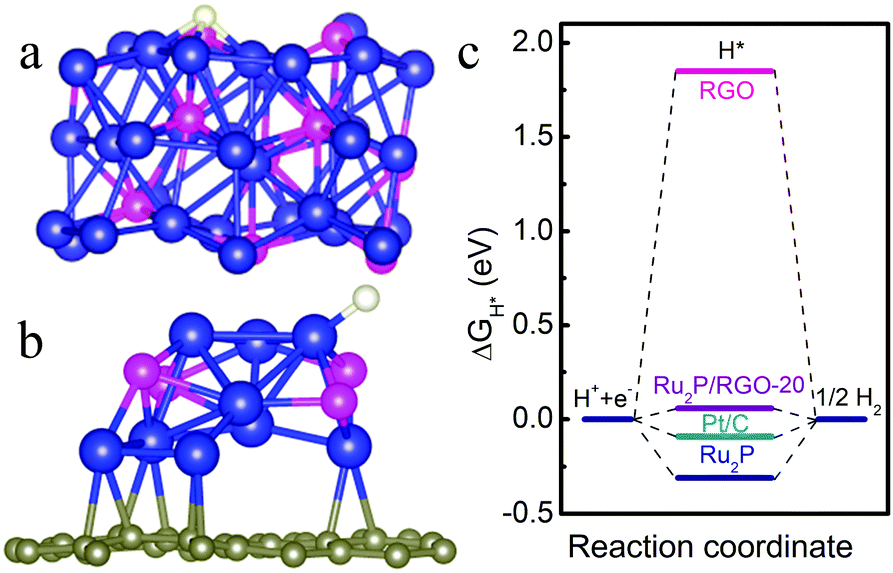
DFT calculation to probe the hydrogen adsorption energies on the active catalytic surface of the Ru2P catalyst. a) and b) front and side views of the calculated Ru2P/reduced graphene oxide surface. c) free energy diagram for the HER with different catalysts.
The electrocatalyst is comprised of small, uniform Ru2P nanoparticles (~2-4 nm) evenly distributed on reduced graphene oxide sheets. The activity of the prepared catalyst (1.0 mg cm-2) for the HER was measured in an acidic medium (0.5 M H2SO4) and the overpotential to achieve a current density of -10 mA cm-2 was -22 mV, superior to Pt/C (-27 mV). In an alkaline environment (1.0 M KOH) catalyst performance was enhanced, with an overpotential of -13 mV (29 mV lower than Pt/C). High Faradaic efficiencies of more than 98% were measured in both acidic and alkaline solutions. Additionally, analysis was undertaken to further understand how the structure and composition of the catalyst influences its activity. Double layer capacitance measurements gave clues about the catalyst surface, while theoretical DFT calculations were used to study H-adsorption energies.
There is no way to avoid the reality that ruthenium is also a rare and costly metal, and for this reason may not hold the key to solving our energy woes. However, of real value are the insights gained from probing the structure function relationship of this highly active catalyst, which may guide the synthesis of rationally-designed catalysts using inexpensive and abundant materials.
To find out more please read:
Ultrasmall Ru2P nanoparticles on graphene: a highly efficient hydrogen evolution reaction electrocatalyst in both acidic and alkaline media
Tingting Liu, Shuo Wang, Qiuju Zhang, Liang Chen, Weihua Hu, Chang Ming Li.
Chem. Commun., 2018, 54, 3343-3346
DOI: 10.1039/c8cc01166d
 About the author:
About the author:
Zoë Hearne is a PhD candidate in chemistry at McGill University in Montréal, Canada, under the supervision of Professor Chao-Jun Li. She hails from Canberra, Australia, where she completed her undergraduate degree. Her current research focuses on transition metal catalysis to effect novel transformations, and out of the lab she is an enthusiastic chemistry tutor and science communicator.










")



![Upon photoexcitation of [Ru(bpy)3]2+, electron transfer through a ferredoxin scaffold to a cobaloxime catalyst facilitates the production of hydrogen.](http://pubs.rsc.org/services/images/RSCpubs.ePlatform.Service.FreeContent.ImageService.svc/ImageService/image/GA?id=C5CC03006D) It is the latter approach which
It is the latter approach which 

{kind=link}