Proteins have an expansive utility in the structure, function, replication and regulation of all cells, and developing tools to study each role is to the benefit of our continued health and wellbeing. One tool is protein bioconjugation, the covalent pairing of a molecule with a protein. Molecule-protein combinations are endless, provided there are efficient methods available to couple molecules with amino acids. Among bioconjugation methods, cysteine functionalisation is a popular choice because the primary thiol is highly nucleophilic thus aiding chemoselectivity. Furthermore, cysteine is rare, reducing the likelihood of many competing, reactive residues.
Transition metal catalysed transformations are uncommon in bioconjugations, despite prominence in other areas of synthetic chemistry. This is because only the most robust methods can overcome the challenges of this chemistry: the solubility of substrates in solvents other than aqueous media, the presence of other amino acids bearing reactive functional groups, and the requirement for low temperatures, low concentrations and mild pH to preserve protein structure.
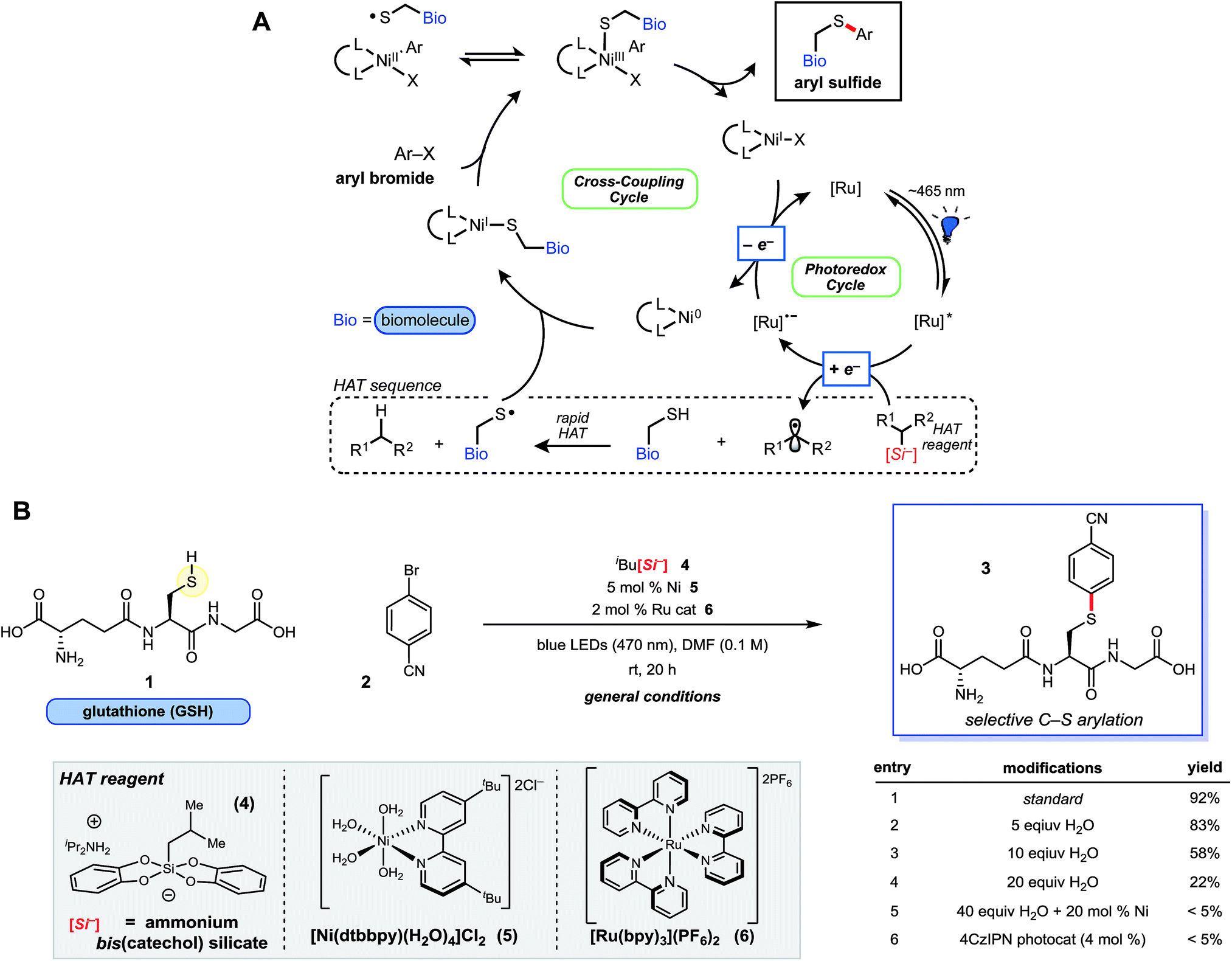
Catalytic cycle for the nickel/phororedox catalysed synthesis of cysteine bioconjugates
A group of researchers from the University of Pennsylvania headed by Professor Gary Molander have developed a bioconjugation method in which aryl halides are cross-coupled with cysteine residues in peptides. Two complexes catalyse the reaction in two connected cycles: the photoredox cycle by a ruthenium-bipyridine complex, and the catalytic cycle by a nickel-bipyridine complex.
The reaction is efficient at room temperature and does not require prior functional group protection. The reaction can also be performed under dilute conditions (10 mM) and on gram scale (3.5 mmol). The scope table includes more than 35 reactions coupling a broad range of aryl halides with small peptides (4 and 9 amino acids) and biologically relevant molecules such as coenzyme A and sulphur-containing pharmaceuticals.
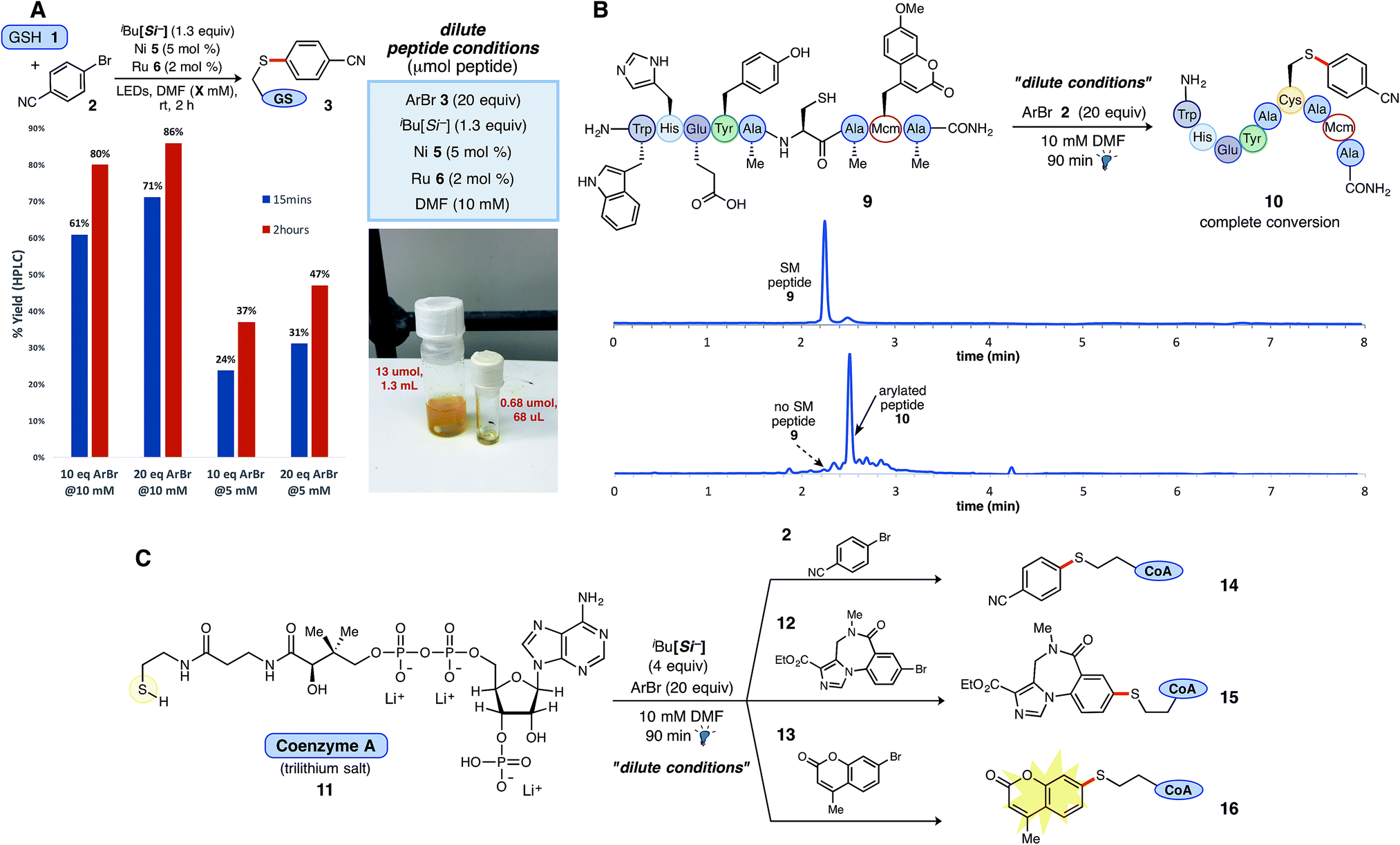
Protecting group free functionalisation of small peptides under dilute conditions
Included in the reaction scope are a number of substrates which highlight how this work can adapt to established techniques for studying proteins. Coupling of a coumarin generates a fluorescent molecule, which could be used to study the cellular localisation of a protein. Reaction with an aryl-bound biotin derivative demonstrates that affinity tags can be coupled, and utilising aryl-containing pharmaceutical agents is relevant to the synthesis of antibody-drug conjugates.
With this research the authors have contributed a robust catalytic system, which convincingly shows the value of combining a transition metal and photoredox catalyst to functionalise cysteine residues in biomolecules. A necessary next step for this chemistry, and no small task, is to further optimise the reaction conditions for whole proteins.
Read the research article:
Scalable thioarylation of unprotected peptides and biomolecules under Ni/photoredox catalysis
Chem. Sci., 2018, DOI: 10.1039/C7SC04292B
Brandon A. Vara, Xingpin Li, Simon Berritt, Christopher R. Walters, E. James Petersson, Gary A. Molander.
 About the Author:
About the Author:
Zoë Hearne is a PhD candidate in chemistry at McGill University in Montréal, Canada, under the supervision of Professor Chao-Jun Li. She hails from Canberra, Australia, where she completed her undergraduate degree. Her current research focuses on transition metal catalysis to effect novel transformations, and out of the lab she is an enthusiastic chemistry tutor and science communicator.








")