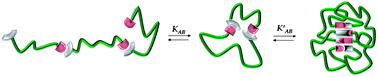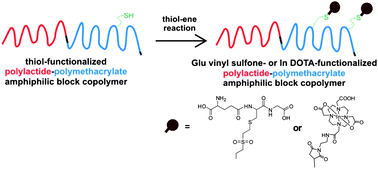
In this publication, Themistou, Battaglia and Armes reported on the one-pot metal-free ring-opening polymerization (ROP)–reversible addition–fragmentation chain transfer (RAFT) synthesis of biocompatible linear and branched amphiphilic diblock copolymers based on a biodegradable aliphatic polyester (PLA) and methacrylic monomers (such as 2-(dimethylamino)ethyl methacrylate (DMA) or oligo(ethylene glycol) methacrylate (OEGMA)), using a novel hydroxyl-functionalized trithiocarbonate-based chain transfer agent. These amphiphilic diblock copolymers self-assembled in dilute aqueous solution, leading to various copolymer morphologies depending on the block compositions. Two novel disulfide-functionalized PLA-branched block copolymers were also synthesized using simultaneous ROP of LA and RAFT copolymerization of OEGMA or DMA with a disulfide-based dimethacrylate. The disulfide bonds were reductively cleaved using tributyl phosphine to generate reactive thiol groups. Thiol–ene chemistry was utilized for further derivatization with thiol-based biologically important molecules and heavy metals for tissue engineering or bioimaging applications, respectively.
Facile synthesis of thiol-functionalized amphiphilic polylactide–methacrylic diblock copolymers by Efrosyni Themistou, Giuseppe Battaglia and Steven P. Armes Polym. Chem. 2014, 5, 1405-1417.
Julien Nicolas is a web-writer and advisory board member for Polymer Chemistry. He currently works at Univ. Paris-Sud (FR) as a CNRS researcher.








")