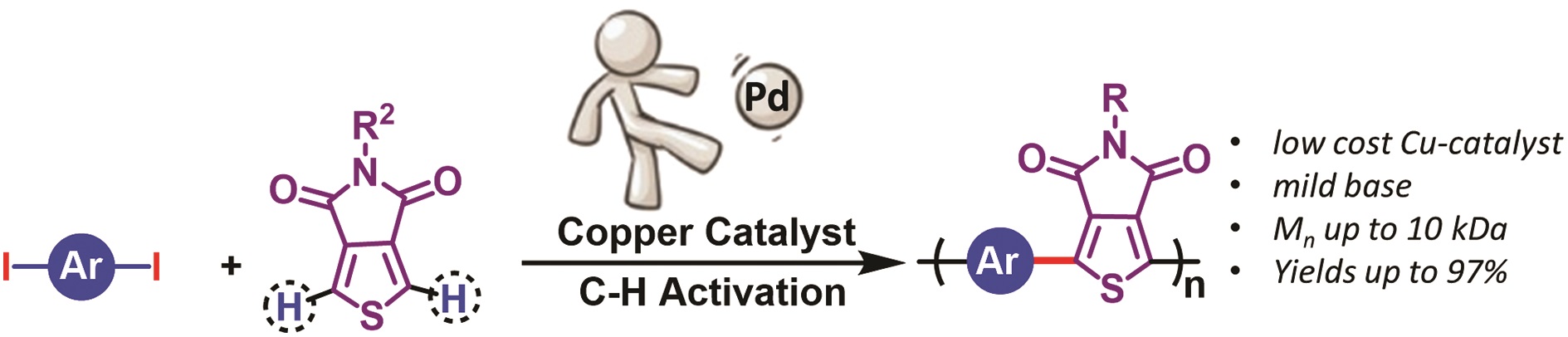
Thermoresponsive polymers can be used in a wide range of applications ranging from drug delivery to bioengineering owing to their unique capability of undergoing a soluble-to-insoluble transition in response to an external thermal stimulus. Amphiphilic block copolymers that contain a thermoresponsive block can self-assemble into core-corona nanoassemblies where the core consists of the hydrophobic block and the corona is formed by the thermoresponsive block. Zhang, Han and co-workers were interested in studying the dependence of a thermoresponsive phase transition on the topology structure. To achieve this, reversible addition-fragmentation chain transfer (RAFT) dispersion polymerization was employed to synthesize well-defined multi-arm star block copolymer nanoassemblies via polymerization-induced self-assembly. The block copolymer was designed to have the first part consisting of the thermoresponsive poly(N-isopropylacrylamide) (PNIPAM) and the second block being the hydrophobic polystyrene (PS). By carefully modifying the number of arms (n=1, 2, 3 and 4), the degree of polymerization of the hydrophobic block and the polymerization conditions, (PNIPAM-b-PS) nanoassemblies with similar degree of polymerization and chain density, albeit different topology structure, were obtained. A range of characterization techniques were subsequently employed to comparatively study the responsiveness of these materials including turbidity analysis, dynamic light scattering, variable-temperature 1H NMR and rheological analysis. The authors found that the topology of the tethered PNIPAM chains had a significant influence on their thermoresponsive phase transition which decreased upon increasing the number of arms. This can be attributed to the inter-and intra-particle chain entanglement in the synthesized star nanoassemblies. It can thus be concluded that the topology of the thermoresponsive polymers can significantly affect their thermoresponsive and should be taken into account when designing the synthesis of such materials.
This paper is FREE to read and download until 27th February!
Synthesis of star thermoresponsive amphiphilic block copolymer nano-assemblies and the effect of topology on their thermoresponse, Polym. Chem., 2019, 10, 403-411, DOI: 10.1039/C8PY01617H
About the Webwriter
 Dr. Athina Anastasaki is an Editorial Board Member and a Web Writer for Polymer Chemistry. Since January 2019, she joined the Materials Department of ETH Zurich as an Assistant Professor to establish her independent research group.
Dr. Athina Anastasaki is an Editorial Board Member and a Web Writer for Polymer Chemistry. Since January 2019, she joined the Materials Department of ETH Zurich as an Assistant Professor to establish her independent research group.








")