A concise synthesis of the natural product rasfonin could reignite interest in this molecule as a tool to develop cancer drugs, say scientists from the Netherlands.
Adriaan Minnaard and Ben Feringa’s group from the University of Groningen developed the synthesis, which has a higher overall yield and takes fewer steps than previous syntheses, they say.
Rasfonin could be important in cancer research because it inhibits a cell signalling pathway called the RAS pathway. ‘If a cell becomes cancerous, often you see that RAS, the most important protein in the pathway, is mutated,’ explains Minnaard. The mutated RAS keeps on signalling, so the cancer cells keep dividing. Rasfonin blocks the pathway by binding either to RAS or one of the other proteins in the pathway, but it’s not known which one. ‘Scientists would love to have rasfonin to investigate this further, but it takes too much time and effort to make,’ says Minnaard.
In a bid to make synthesis easier, the team has made rasfonin in 30 steps (21 in the longest linear sequence) and in 11 per cent overall yield. ‘It’s quite a lot of steps,’ admits Minnaard, ‘but given the overall yield and the good activity (which means that not so much compound is needed), the synthesis is suitable to prepare any quantity needed for research purposes.’ One main step was introducing the methyl groups in the upper chain of rasfonin using catalytic asymmetric conjugate addition and the other was forming the lower chain, which involved using Feringa butenolide, a building block developed by Feringa. ‘It makes our synthesis much more straightforward than the current two syntheses,’ says Minnaard.
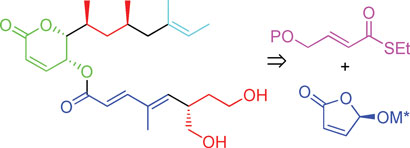
The synthesis of rasfonin was developed using asymmetric conjugate addition and Feringa's butenolide
The first synthesis in 2003 elucidated the stereochemistry and structure of rasfonin, but was not aimed at creating a synthesis for producing large quantities. Masami Ishibashi from Chiba University, Japan, who carried out this synthesis, says that Minnaard and Feringa have achieved a highly stereoselective and efficient synthesis. ‘It may be strongly expected that this new route will accelerate the chemical biology of rasfonin to identify its target protein,’ says Ishibashi.
‘It is a nice piece of work, which highlights some of the asymmetric catalysis methods developed in Professor Feringa’s laboratory,’ adds Robert Boeckman from the University of Rochester, US, who carried out the second synthesis of rasfonin in 2006. Boeckman’s synthesis used chiral auxiliaries. ‘Boeckman changes the substrate in such a way that the stereochemical outcome will be the desired one, but he has to do a lot more steps with the substrates,’says Minnaard. ‘We have our information in the catalyst, which is more efficient.’
Interested? Read Elinor Richards’ full Chemistry World article here or download the OBC paper:
A concise asymmetric synthesis of (−)-rasfonin
Yange Huang, Adriaan J. Minnaard and Ben L. Feringa
Org. Biomol. Chem., 2012, Advance Article
DOI: 10.1039/C1OB06700A









")








