In previous posts, we have discussed the automated workflow we use to check new incoming data for structure and synonym errors. These checks allow us to remove the most common types of errors before they are added to the site. However, these filters do not apply to data already in ChemSpider.
Manual curation is an important part of our work. We periodically review the data on our most accessed records, in addition to ad-hoc removal or correction of erroneous data that we or our users notice when using the site. However, there are far too many records and far too much data to clean up using manual curation alone.
Recently we have focused on bulk identification and removal of erroneous data. This work has covered mapping errors and other clearly incorrect values in our experimental property data, correction or removal of malformed synonyms, correction of incorrectly labelled synonyms, and resolution of structure/synonym clashes.
Experimental Properties
We retrieved all 6.3 million experimental properties, text properties, and associated annotations from the ChemSpider database. We then compared the original text of the property as it was written in the original file to how that text was parsed and mapped by our deposition system. This enabled us to identify and correct several types of errors affecting around 2% of the properties in our database:
- 35,774 experimental property values had been assigned the incorrect unit (e.g. g/L instead of g/mL, °C instead of °F)
- 2,591 boiling points measured under non-standard pressure did not have this pressure displayed
- 4,292 densities had their density and temperature values swapped
- 79,252 miscellaneous erroneous properties and associated annotations were deleted. For example, “white crystals” mapped as melting point, impossibly high melting points or densities, etc.
Synonyms
Synonyms, chemical names, and identifiers are the most abundant type of data on ChemSpider, with a total of more than 446 million synonyms. These synonyms have additional metadata including language labels and flags identifying what type of synonym they are (e.g. CAS number, UNII, INN, trade name).
Simple Checks
We ran a series of regular expression string searches to identify synonyms with incorrect metadata, as well as malformed or otherwise erroneous synonyms.
- 200,007 synonym type flags added, and 4,766 incorrect flags removed
- 9,170 synonyms with an incorrect language label identified.
- 631,697 erroneous synonyms identified, including scrambled characters, properties/units, molecular formulae as synonyms, purity information, or invalid CAS numbers or EC numbers (formerly called EINECS).
- 922,334 instances of these erroneous synonyms deleted from ChemSpider records.
Structure/Synonym comparison
After identifying and removing these synonym-level errors, we then cross-checked ChemSpider records and their synonyms to identify mismatches. This work included amino acids, nucleic acids, and pharmaceutically acceptable salts.
As a first pass, we compared synonyms to molecular formulae to identify records missing key elements. Examples include synonyms describing a sodium salt when the molecular formula does not contain sodium, or describing an amino acid when the molecular formula contains no nitrogen. A total of 28,194 of these synonym/formula clashes were identified and removed.
For records that passed this initial molecular formula check, we performed a SMARTS comparison to identify chemical structures missing key structural features described in the synonym. These SMARTS strings were written broadly, with common substitutions allowed to prevent unnecessary removal of valid synonyms from derivative compounds.
In the following examples, the mismatched part of the synonym is highlighted in bold.
Structure
|
Removed synonym
|
 |
Sulfate ion |
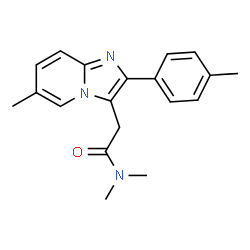 |
Zolpidem tartrate |
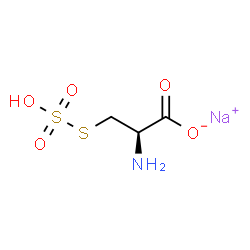 |
Sodium S-sulfocysteine hydrate |
After identifying these clashes, we manually spot-checked the output to weed out false positives and iterate the SMARTS filters. 101,257 synonym/structure clashes were identified and removed.
These checks included the following categories:
- Amino acids and their derivatives: 6 formula clashes, 56 structure clashes
- Nucleic acids, nucleosides, nucleotides: 977 formula clashes, 1,870 structure clashes
- Halogens: 13,437 formula clashes, 1,256 structure clashes
- Alkali and alkaline earth metals, and aluminium: 3,586 formula clashes, 56 structure clashes
- Carboxylic acids and their derivatives: 5,002 formula clashes, 88,501 structure clashes
- Other pharmaceutically acceptable acids: 3,534 formula clashes, 1,529 structure clashes
- Amides and amines: 190 formula clashes, 304 structure clashes
- Deuterates, hydrates, methylbromides: 1,462 formula clashes, 7,685 structure clashes
Get involved
You are the expert in your area of chemistry, so if you see something that doesn’t look quite right please let us know. If the error is confined to a single ChemSpider record, click the “Comment On This Record” box at the top of the affected record and let us know what the problem is. All we need is a sentence describing the error, however the more information you can provide, the better.
For more systemic errors, or in cases where you want to attach supplementary information or corrected chemical structures, please get in touch via email (chemspider@rsc.org).
Comments Off on ChemSpider data cleanup

 Dr. Oliver Koepler is head of the Lab Linked Scientific Knowledge at TIB – Leibniz Information Centre for Science and Technology, where he leads research and developments at the intersection of chemistry, data science, and knowledge engineering. Since 2005, he has been instrumental in transforming chemical research through digital innovation, developing e-infrastructures and research data management services that support scientists throughout the entire data lifecycle. He is a spokesperson of NFDI4Chem which has developed innovative tools like the SmartLab environment with Chemotion electronic lab notebook, the federation of Data repositories, the Terminology and Search Service that promote FAIR data principles across the chemistry community.
Dr. Oliver Koepler is head of the Lab Linked Scientific Knowledge at TIB – Leibniz Information Centre for Science and Technology, where he leads research and developments at the intersection of chemistry, data science, and knowledge engineering. Since 2005, he has been instrumental in transforming chemical research through digital innovation, developing e-infrastructures and research data management services that support scientists throughout the entire data lifecycle. He is a spokesperson of NFDI4Chem which has developed innovative tools like the SmartLab environment with Chemotion electronic lab notebook, the federation of Data repositories, the Terminology and Search Service that promote FAIR data principles across the chemistry community. Jonathan Hirst is Professor in Computational Chemistry at the University of Nottingham. In 2020, he was awarded a Chair in Emerging Technologies by the Royal Academy of Engineering, focusing on research that will empower the development of next-generation molecules that chemical engineers and chemists make, by using machine learning to augment human decision-making. His tenure as Head of School (2013-2017) saw some significant transformations under his leadership, including the building of the GSK Carbon Neutral Laboratory and a successful bid for an Athena Swan Silver Award.
Jonathan Hirst is Professor in Computational Chemistry at the University of Nottingham. In 2020, he was awarded a Chair in Emerging Technologies by the Royal Academy of Engineering, focusing on research that will empower the development of next-generation molecules that chemical engineers and chemists make, by using machine learning to augment human decision-making. His tenure as Head of School (2013-2017) saw some significant transformations under his leadership, including the building of the GSK Carbon Neutral Laboratory and a successful bid for an Athena Swan Silver Award. Nicolas
Nicolas 

 Wendy Patterson is a member of the Board of Management of the Beilstein-Institut where she serves as Scientific Director. She is also a board member of Crossref and the InChI Trust, in addition to other non-profit organizations serving the community. The Beilstein-Institut supports the scientific community in communicating and disseminating high-quality information in chemistry and related sciences through open science practices. In addition, they support the scientific community by funding educational and cultural projects. The Beilstein-Institut recently established the Beilstein ChemInfo Labs, a project area supporting community-led digital infrastructure and standards projects in chemistry.
Wendy Patterson is a member of the Board of Management of the Beilstein-Institut where she serves as Scientific Director. She is also a board member of Crossref and the InChI Trust, in addition to other non-profit organizations serving the community. The Beilstein-Institut supports the scientific community in communicating and disseminating high-quality information in chemistry and related sciences through open science practices. In addition, they support the scientific community by funding educational and cultural projects. The Beilstein-Institut recently established the Beilstein ChemInfo Labs, a project area supporting community-led digital infrastructure and standards projects in chemistry. Dr. Jacob Al-Saleem, a Data Science Manager at CAS, a division of the American Chemical Society, leads a team working to develop novel bioinformatic solutions using artificial intelligence to extract and connect scientific knowledge. He led the data science efforts for a team that developed a heterogeneous knowledge graph-based approach to identify repositionable therapeutics for COVID-19. His knowledge graph work is the foundation for the Life Sciences Knowledge Graph that is featured in CAS BioFinder. Dr. Al-Saleem earned his B.S. in Molecular Genetics and Ph.D. in Molecular Cellular and Developmental Biology from The Ohio State University, where his work focused on the molecular virology of Human T cell Leukemia Viruses.
Dr. Jacob Al-Saleem, a Data Science Manager at CAS, a division of the American Chemical Society, leads a team working to develop novel bioinformatic solutions using artificial intelligence to extract and connect scientific knowledge. He led the data science efforts for a team that developed a heterogeneous knowledge graph-based approach to identify repositionable therapeutics for COVID-19. His knowledge graph work is the foundation for the Life Sciences Knowledge Graph that is featured in CAS BioFinder. Dr. Al-Saleem earned his B.S. in Molecular Genetics and Ph.D. in Molecular Cellular and Developmental Biology from The Ohio State University, where his work focused on the molecular virology of Human T cell Leukemia Viruses. Birgit (Bea) Braun, Ph.D., is an R&D/TS&D Fellow for Digital Innovation in the Packaging, Specialty Plastics & Hydrocarbons R&D organization at Dow. She has held diverse roles across R&D and M&E, and for more than a decade, Bea has focused on applying Artificial Intelligence and Machine Learning to chemical manufacturing and materials research. She has led the development and deployment of numerous data science solutions across Dow, and today her work centers on unlocking value through holistic digital transformation. Bea earned a Dipl.-Ing. in Process Engineering with a specialization in Industrial Environmental Protection from Montanuniversität Leoben in Austria, she holds an M.S. in Environmental Science & Engineering and a Ph.D. in Chemical Engineering from the Colorado School of Mines.
Birgit (Bea) Braun, Ph.D., is an R&D/TS&D Fellow for Digital Innovation in the Packaging, Specialty Plastics & Hydrocarbons R&D organization at Dow. She has held diverse roles across R&D and M&E, and for more than a decade, Bea has focused on applying Artificial Intelligence and Machine Learning to chemical manufacturing and materials research. She has led the development and deployment of numerous data science solutions across Dow, and today her work centers on unlocking value through holistic digital transformation. Bea earned a Dipl.-Ing. in Process Engineering with a specialization in Industrial Environmental Protection from Montanuniversität Leoben in Austria, she holds an M.S. in Environmental Science & Engineering and a Ph.D. in Chemical Engineering from the Colorado School of Mines. Garrison W. (Gary) Cottrell is a Professor of Computer Science and Engineering at UC San Diego. He co-founded the Perceptual Expertise Network, and directed the Temporal Dynamics of Learning Center, an NSF-sponsored Science of Learning Center spread across four countries. Gary’s research is strongly interdisciplinary. He is interested in Cognitive Science and Computational Cognitive Neuroscience and in applying AI to problems in other areas of science or engineering. Most recently he has been using deep learning to elucidate the structure of small (natural product) molecules from their NMR spectra in collaboration with Bill Gerwick at the Scripps Institute of Oceanography. He received his PhD in 1985 from the University of Rochester and did a postdoc at the Institute of Cognitive Science at UCSD until 1987, when he joined the CSE Department.
Garrison W. (Gary) Cottrell is a Professor of Computer Science and Engineering at UC San Diego. He co-founded the Perceptual Expertise Network, and directed the Temporal Dynamics of Learning Center, an NSF-sponsored Science of Learning Center spread across four countries. Gary’s research is strongly interdisciplinary. He is interested in Cognitive Science and Computational Cognitive Neuroscience and in applying AI to problems in other areas of science or engineering. Most recently he has been using deep learning to elucidate the structure of small (natural product) molecules from their NMR spectra in collaboration with Bill Gerwick at the Scripps Institute of Oceanography. He received his PhD in 1985 from the University of Rochester and did a postdoc at the Institute of Cognitive Science at UCSD until 1987, when he joined the CSE Department.















