Tasmania is a well-known touristic destination in Australia. During 1993-1995, Blackman and coworkers reported eleven new alkaloids named Cylindricines A – K (Figure 1) isolated from ascidian Clovelina cylindrical, which were collected from different locations of the East Coast of Tasmania. This group of alkaloids possess new tricyclic ring systems containing an a-tert-alkylamine motif, which are structurally related to lepadiformine and fasicularin.

Figure 1. Structure of representative alkaloids of the cylindricine/fasicularin/lepadiformine family
The intriguing structures of cylindricines have attracted considerable attention from synthetic community. More than twenty research groups including those of Snider, Weinreb, Heathcock, Molander, Kibayashi, Trost, Ciufolini, Hsung, Shibasaki, Donohoe, Renaud, Pandey, and Chida/Sato have been engaged in their total syntheses, which resulted in many elegant approaches. However, efficient and flexible enantioselective synthetic approaches are still demanding.
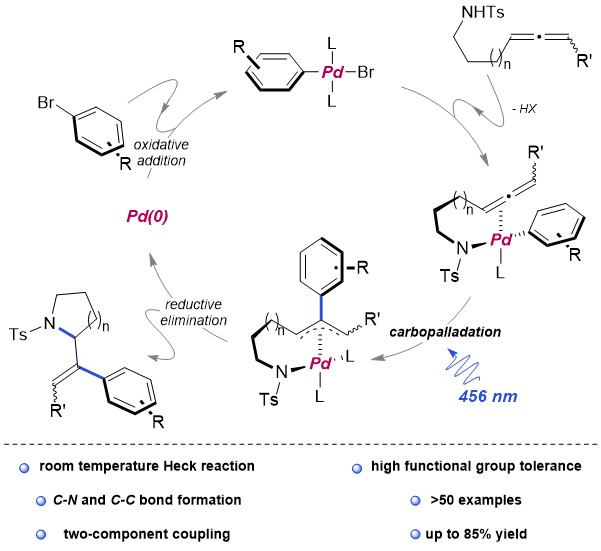
Recently, the group of Huang and collaborators of the Xiamen University have disclosed a concise and versatile enantioselective total syntheses of (+)-cylindricines C, D, E and their diastereomers. Huang’s synthetic strategy to (+)-cylindricines C-E (1c-1e) stemmed from the identification of the a-tert-alkylamine motif-bearing prolinol moiety as the key structural feature and relied on the amides reductive bisalkylation, a versatile reaction originally developed by his group in 2010 and improved by replacing DTBMP with TTBP in 2021 (Scheme 1).
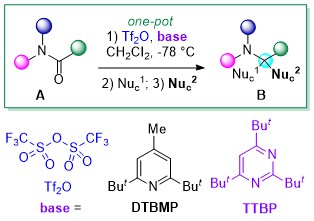
Scheme 1. Huang’s one-pot reductive bisalkylation of amides
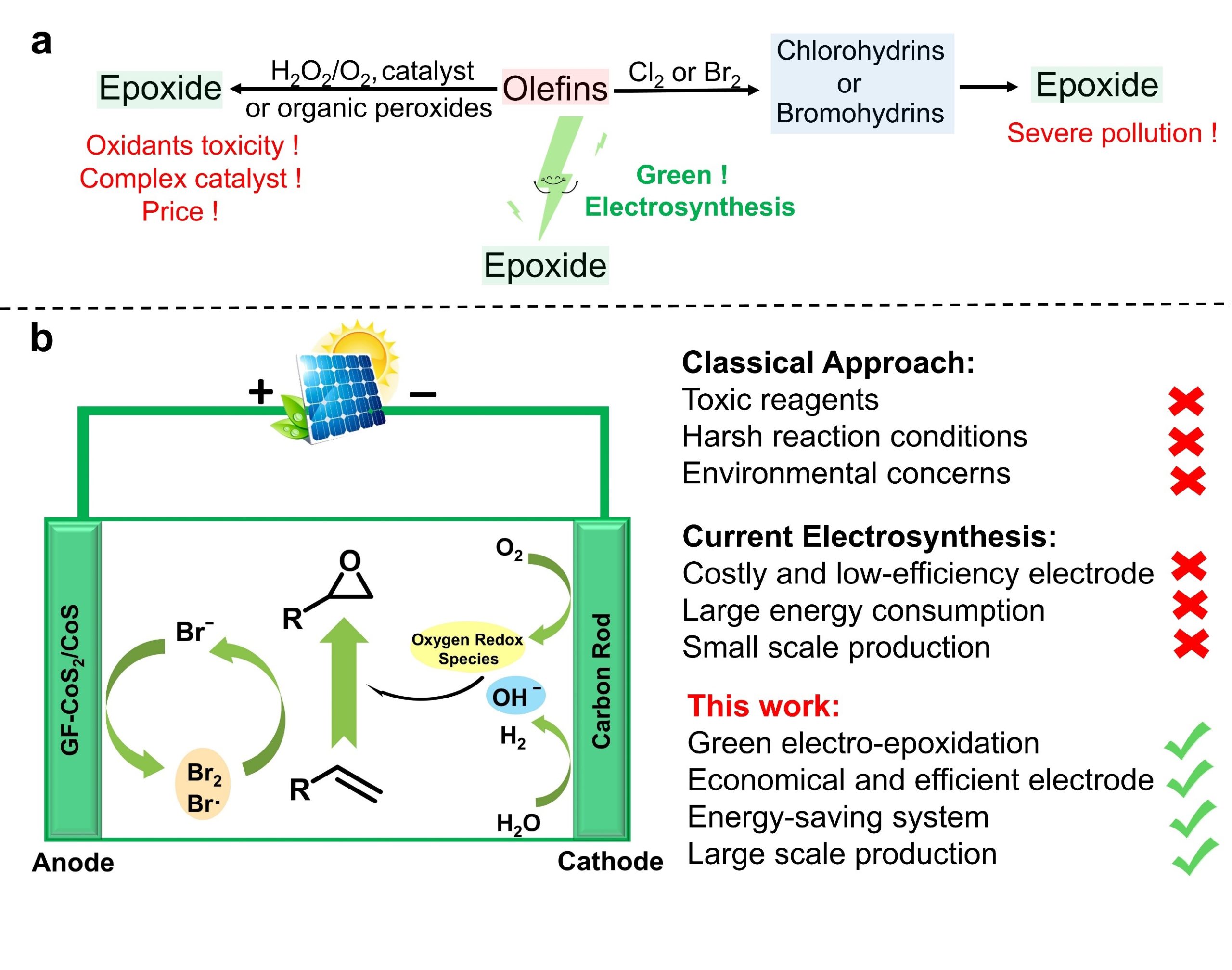
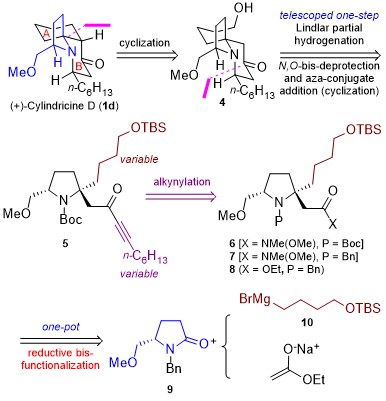
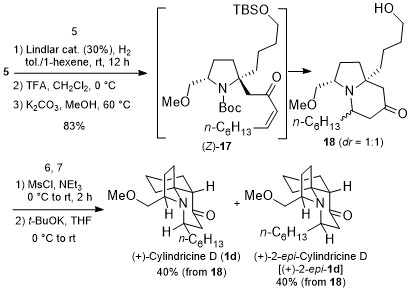
According to the retrosynthetic analysis outlined in Scheme 2, Huang’s enantioselective total synthesis of (+)-cylindricine D (1d) is displayed in Scheme 3. Employing L-pyroglutamic acid derived lactam (S)-9 as the chiral pool, and Tf2O/TTBP (2,4,6-tri-tert-butylpyrimidine) as an amide activation system, the reductive bisalkylation of lactam (S)-9 resulted in the formation of prolinol derivative 8 in 75% yield (dr = 7: 1). Another key feature of the reaction is the telescoping of four reactions [(1) partial reduction of ynone 5 to the presumed (Z)-enone 17; (2) N-deprotection; (3) O-deprotection; (4) intramolecular aza-conjugate addition] into one step, which rendered the synthesis highly efficient. On the other hand, following the same approach, they have synthesized (+)-cylindricines C and E (no shown) via 16.
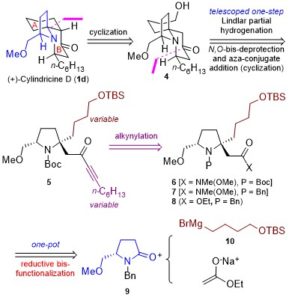
Scheme 2. Huang’s retrosynthetic analysis of (+)-Cylindricine D
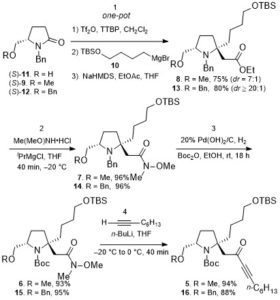
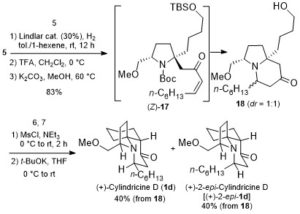
Scheme 3. Enantioselective total synthesis of (+)-cylindricine D (1d)
In summary, they have accomplished one of the shortest and the most efficient total syntheses of (+)-cylindricines C-E so far reported: cylindricine D (1d): 7 steps and 20.9% overall yield from (S)-9; (+)-cylindricine C (1c): 8 steps, 19.1% overall yield from (S)-12; O-Acetylation of (+)-cylindricine C afforded (+)-cylindricine E (1e).
Enantioselective Total Syntheses of Marine Natural Products (+)-Cylindricines C, D, E and Their Diastereomers
Ying-Hong Huang,† Zhan-Jiang Liu,† and Pei-Qiang Huang*

Pei-Qiang Huang obtained his B.Sc. (1982) from Xiamen University (China) and D. E. A. (1984) from Université de Montpellier II (France) under the direction of the late Professor B. Castro (INSERM-CNRS). After accomplishing the research work at the Institut de Chimie des Substances Naturelles (ICSN), CNRS under the supervision of Professor Dr. H.-P. Husson, his received his Ph D from Université de Paris-Sud (Orsay) (France) in 1987. He served as a postdoctoral fellow in the group of the late Professor W.-S. Zhou at Shanghai Institute of Organic Chemistry, CAS in 1988-1990. He was appointed as an associate Professor at Xiamen University in 1990, and was promoted to a full Professor in 1993. Professor Huang’s research team is interested in developing novel and efficient synthetic methodologies, total synthesis of natural products and medicinal relevant molecules, and chemical biology. He has co-editor several books, including: “Efficiency in natural product total synthesis” (Editors: Pei‐Qiang Huang, Zhu‐Jun Yao, Richard P. Hsung; Forwarded by Henry N. C. Wong), John Wiley & Sons, Inc., 2018. He is a fellow of RSC, and currently an associate editor of Org. Chem. Front.
Comments Off on Enantioselective total syntheses of marine natural products (+)-cylindricines C, D, E and their diastereomers











")