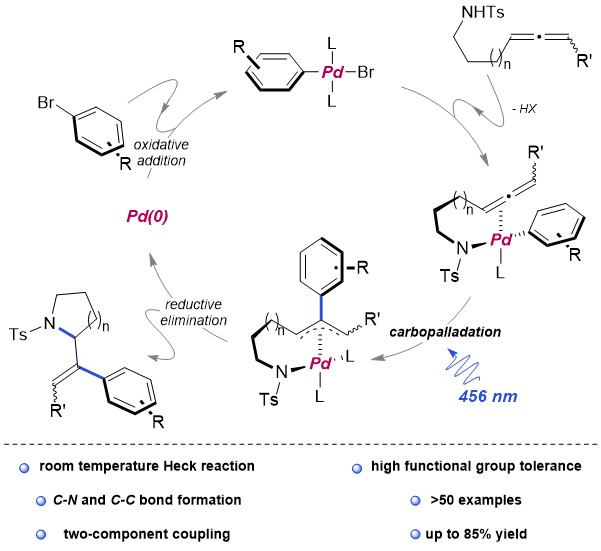
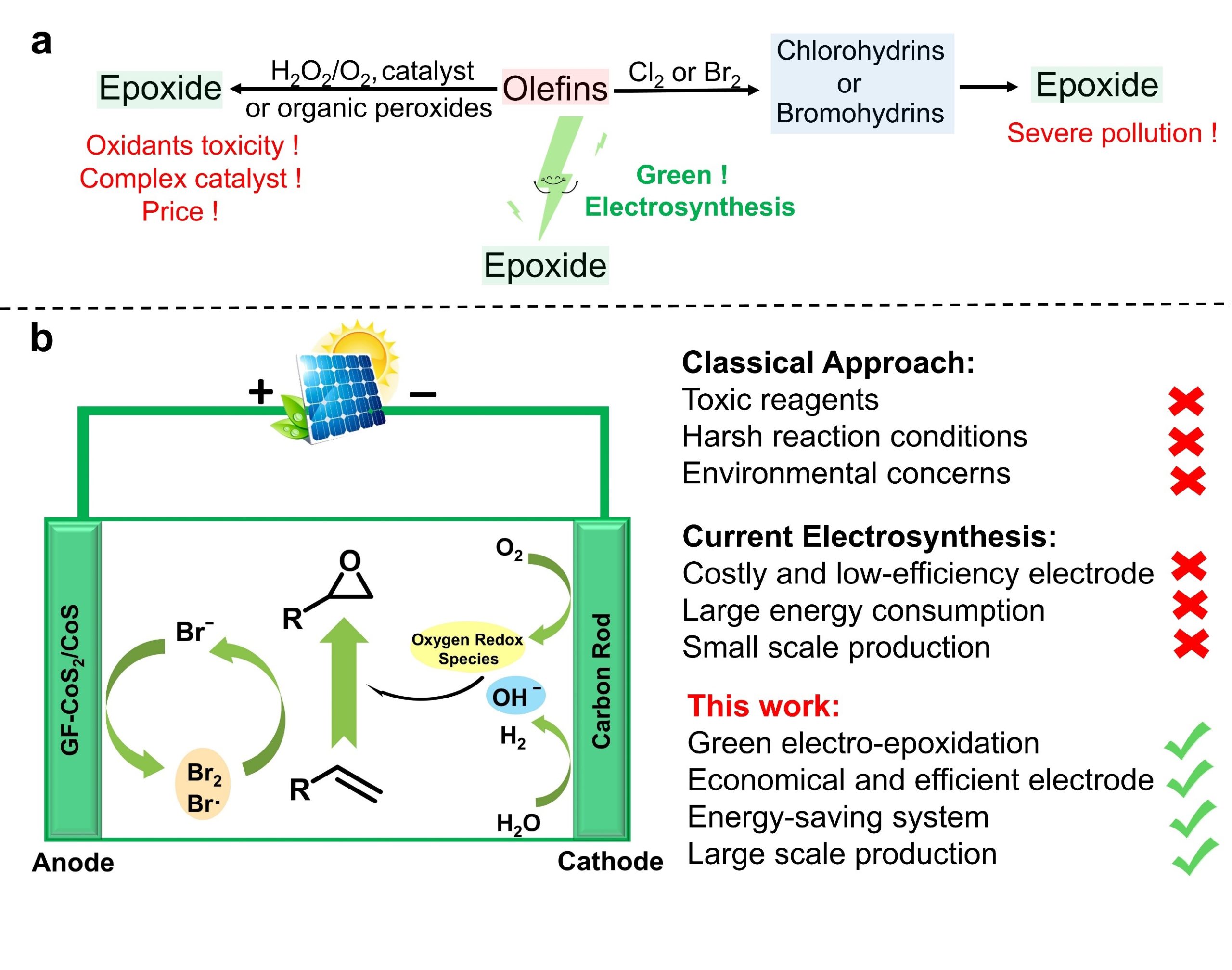
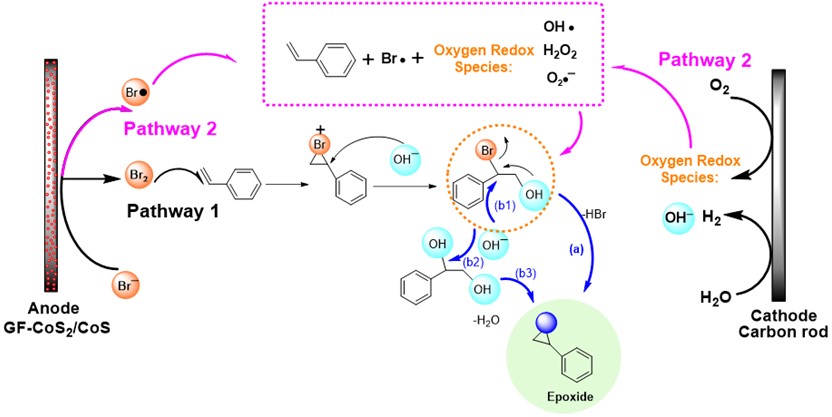
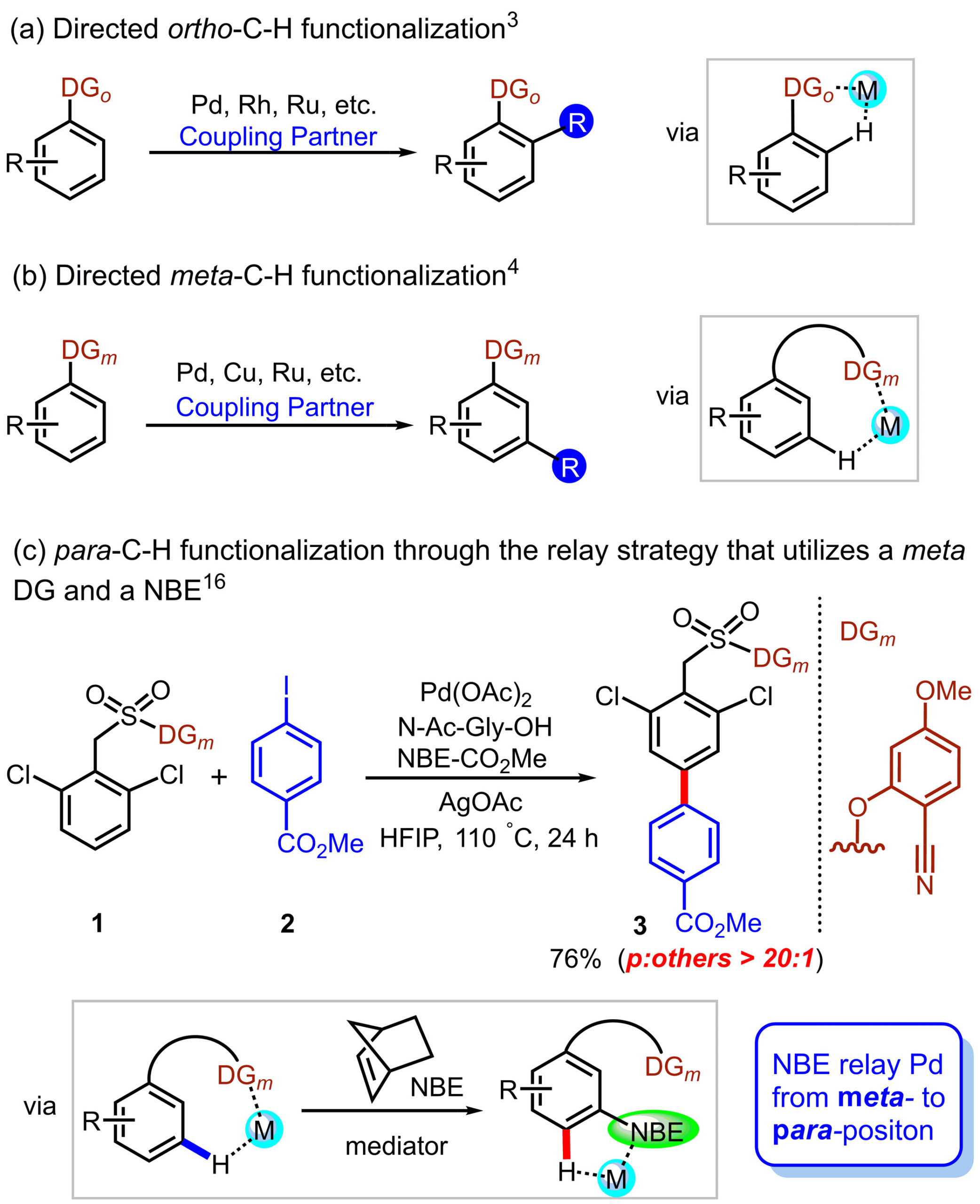
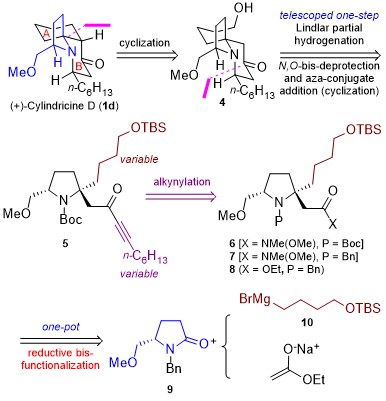
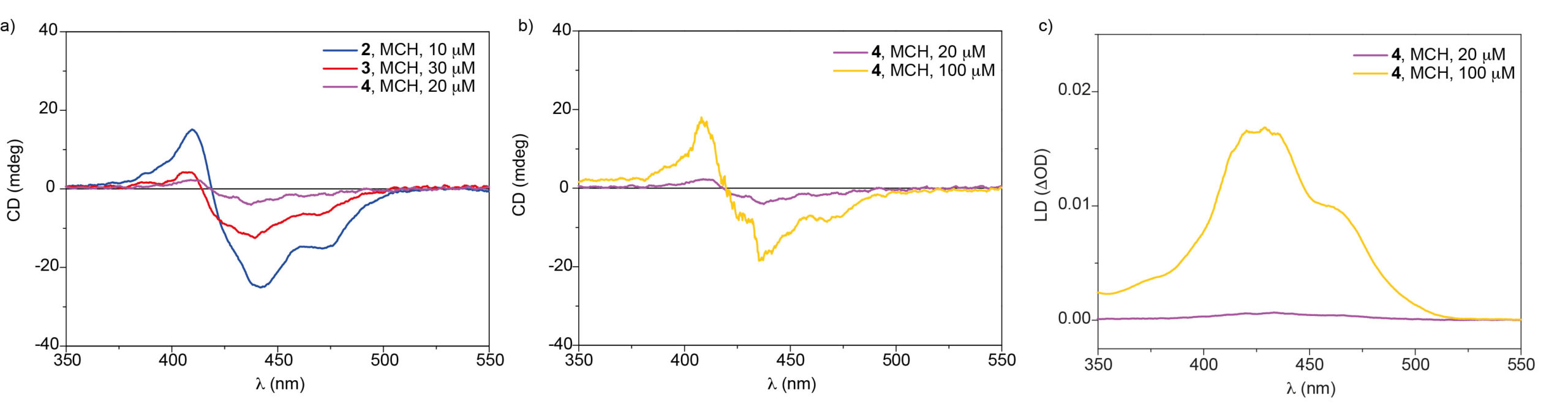
Diborons can directly be transformed into diverse organoboron compounds of high synthetic significance via a variety of transition metal-catalyzed and -free borylation reactions. Besides commonly used symmetrical diborons such as (pin)B–B(pin), recently attention has also been paid to the use of unsymmetrical ones, especially those having different boron-Lewis acidities.
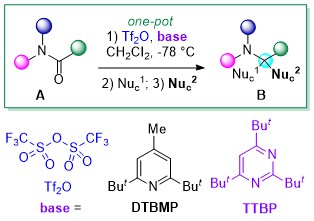
Numerous attention have been directed toward developing unsymmetrical diborons with different boron-Lewis acidities including (pin)B–B(aam) (aam = anthranilamidato), (neop)B–B(dan) (dan = naphthalene-1,8-diaminato) and (pin)B–B(mdan) (mdan = N,N′-dimethyl-naphthalene-1,8-diaminato), and their application in catalytic borylation reactions via σ-bond metathesis as a key elementary step; palladium-catalyzed Miyaura–Ishiyama-type borylation of aryl halides and the copper-catalyzed internal-selective hydroboration of terminal alkynes show the synthetic versatility of (pin)B–B(aam).
Recently, the group of Hiroto Yoshida from Hiroshima University has demonstrated that (pin)B–B(aam) can also be catalytically activated by oxidative addition to a platinum complex, leading to syn-diboration of terminal alkynes, and that a highly electron-deficient triarylphosphine developed by Korenaga, P(BFPy)3, is effective for regiocontrol (Figure 1).

Figure 1 Pt-catalyzed diboration of alkynes with (pin)B–B(aam)
A broad substrate scope of the regioselective diboration has been demonstrated (Figure 2): a variety of aromatic and aliphatic terminal alkynes bearing an electron-donating or electron-withdrawing substituent smoothly underwent the diboration to afford the respective products in high yields without damaging the reactive functionalities. Internal alkynes participating in this reaction also gave syn-products with a high regiocontrol, whereas the reaction of 2-octyne (2x) led to the formation of a mixture of regioisomers (3x and 3′x). Finally, a conjugated diene (4) was also readily transformable into a 1,4-diborated product (5) in a stereoselective fashion.
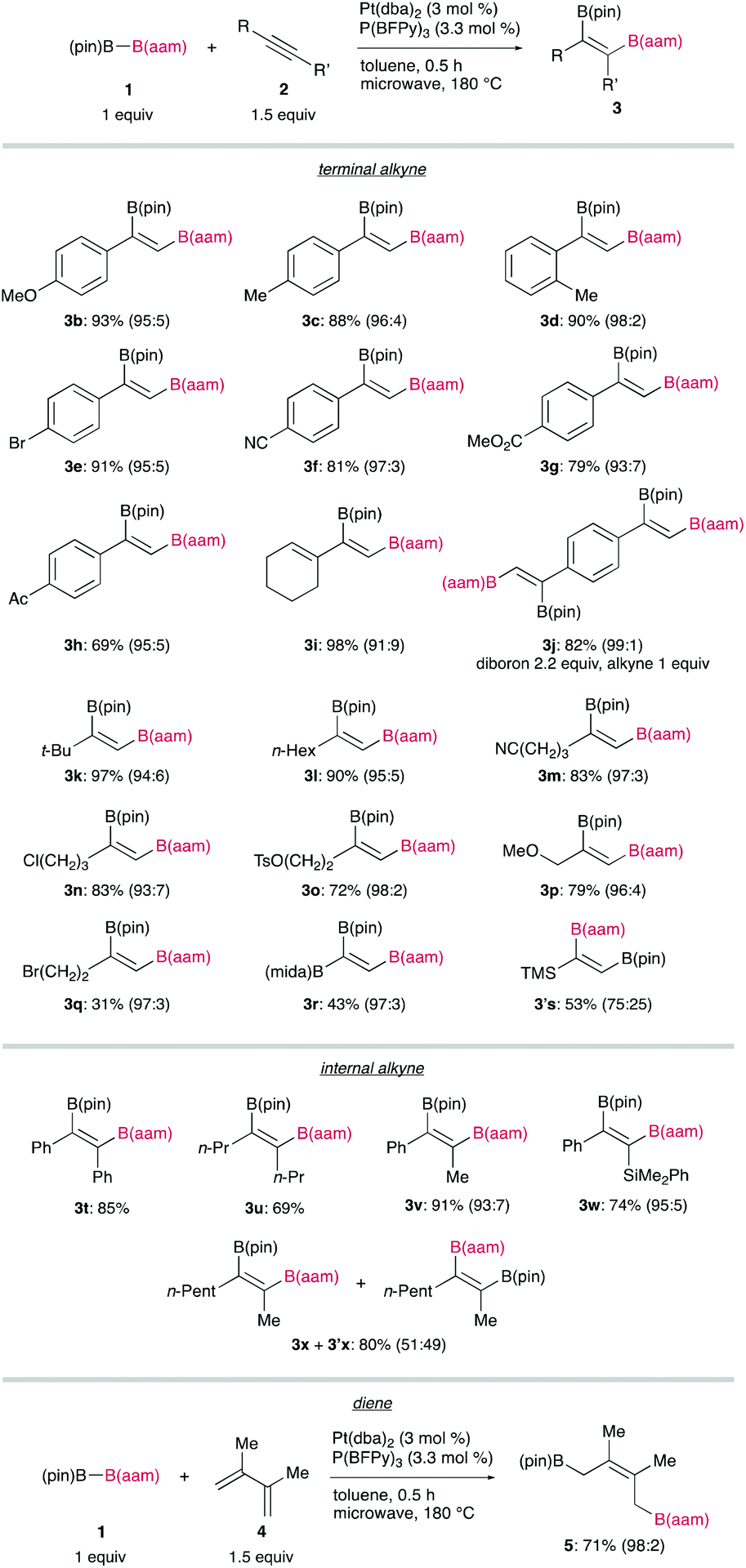
Figure 2 Substrate scope of the diboration
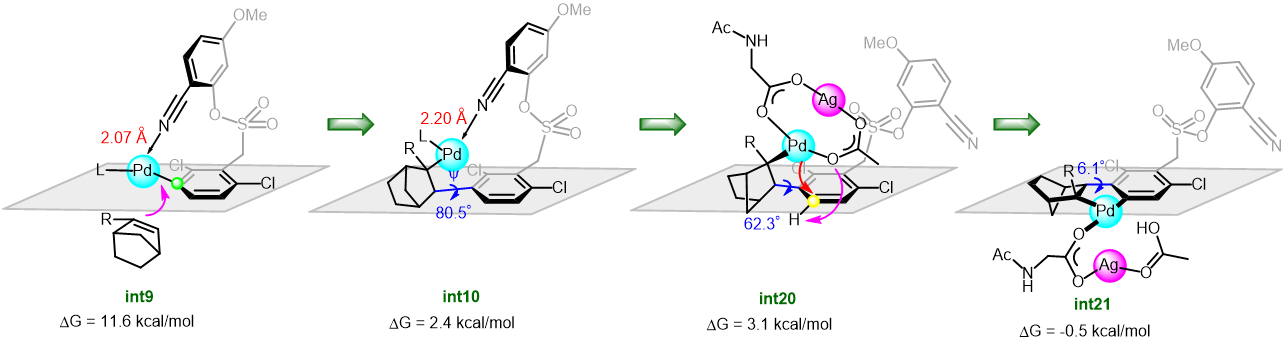
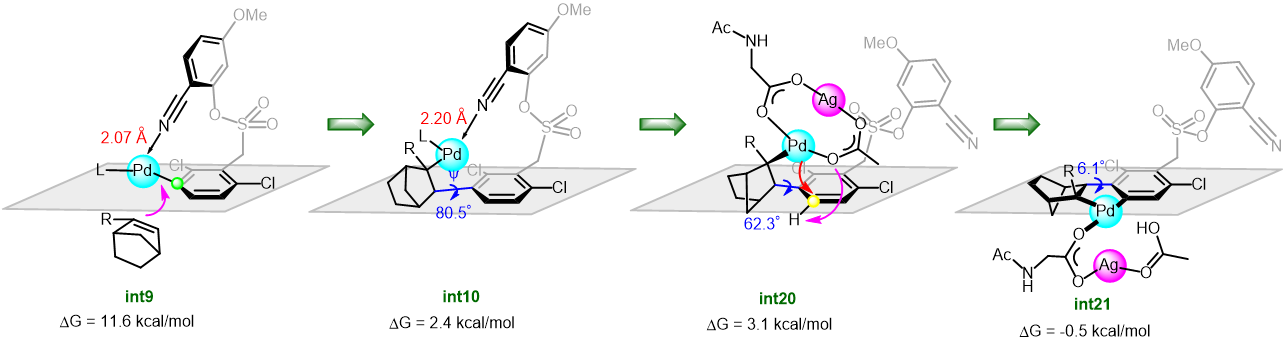
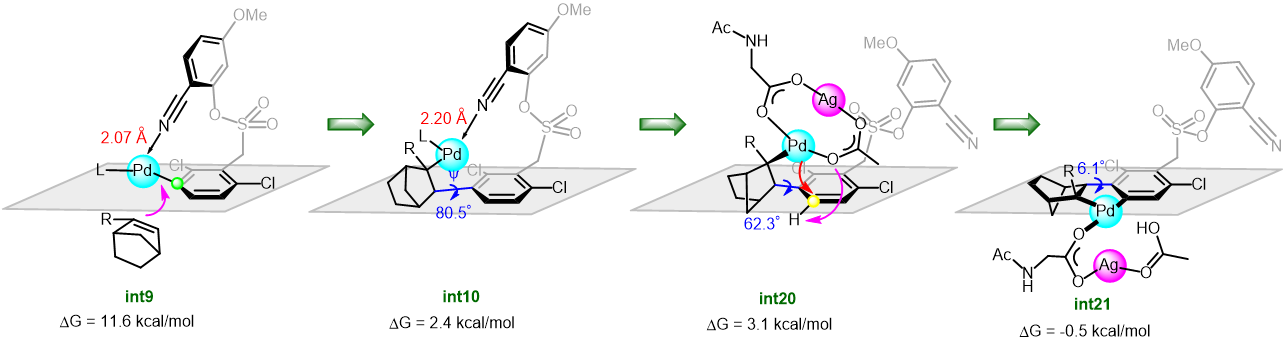
The synthetic utility of the resulting diborylalkene was demonstrated by site-selective cross-coupling to construct a triarylalkene efficiently (Figure 3). In the plausible catalytic cycle, the present diboration could be initiated by the oxidative addition of the boron–boron bond of 1 to a Pt(0) catalyst (Figure 4). The resulting oxidative adduct (8) then accepts the insertion of an alkyne at the Pt–B (pin) bond and/or –B(aam) bond to provide an alkenyl platinum intermediate (9 and/or 9′), which finally affords the diboration product (3) through reductive elimination.
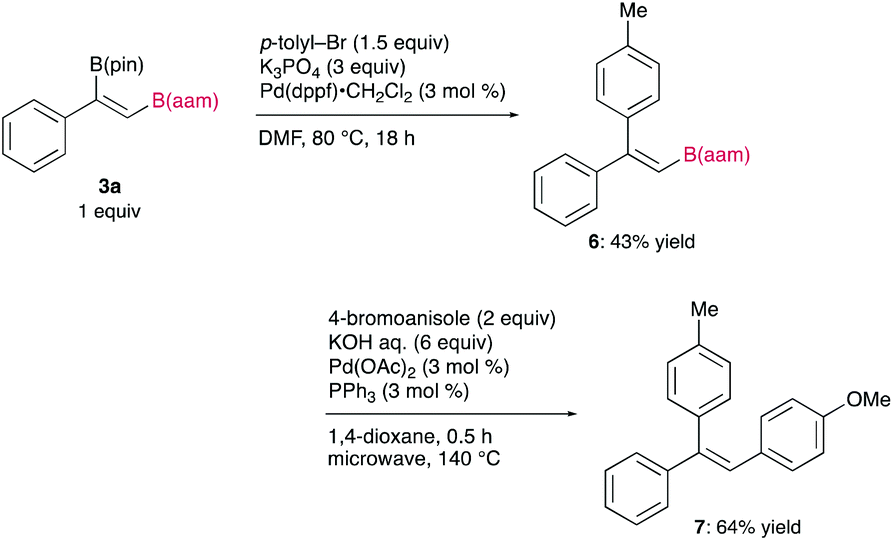
Figure 3 Chemoselective cross-coupling of the diboration product
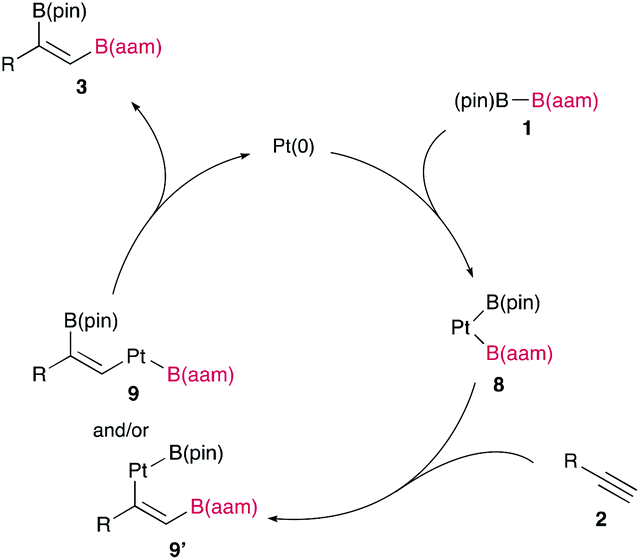
Figure 4 A plausible catalytic cycle for the diboration
(pin)B–B(aam) with different boron-Lewis acidities can be activated by oxidative addition to a platinum(0) complex, leading to regio- and stereo-selective diboration of unsaturated carbon linkages including terminal alkynes, internal alkynes and a conjugated diene. Moreover, the use of a highly electron-deficient triarylphosphine ligand, P(BFPy)3, is indispensable for the regiocontrol, and electron-deficiency in ligands has been proven to be closely correlated with the regioselectivity.
Corresponding Author:

Hiroto Yoshida graduated from Kyoto University in 1996 and received his Ph.D. from Kyoto University under the supervision of Professors Tamejiro Hiyama and Eiji Shirakawa in 2001. He then became an Assistant Professor in 2001 and an Associate Professor in 2006 at Hiroshima University. From 2020, he has been a Full Professor at the same University. His research interests include (1) development of new methods for synthesis of main group organometallics containing boron, tin or silicon, (2) carbon–carbon bond-forming reactions with the main group organometallics, and (3) aryne-based organic synthesis. He is the author of more than 110 articles indexed by SCI and cited more than 5200 times with an index H = 45.
Web of Science ResearcherID: B-7954-2011 (https://publons.com/researcher/1189090/hiroto-yoshida/)








")