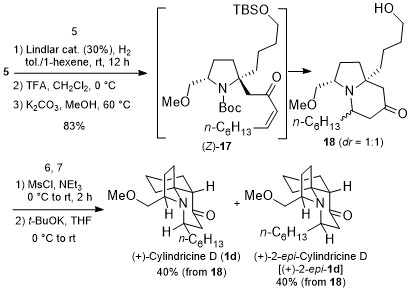
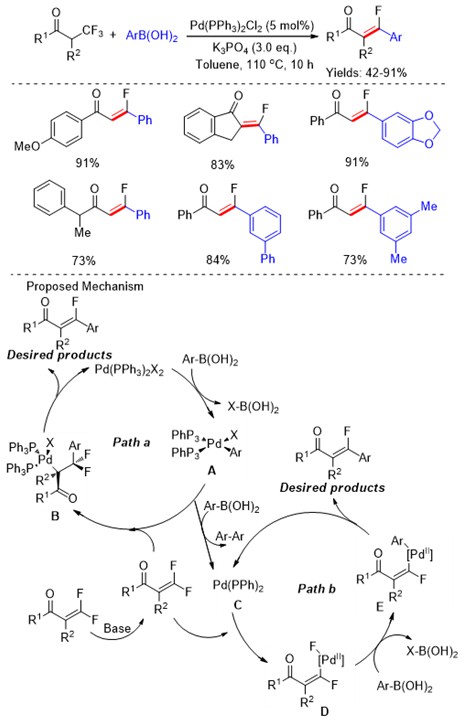
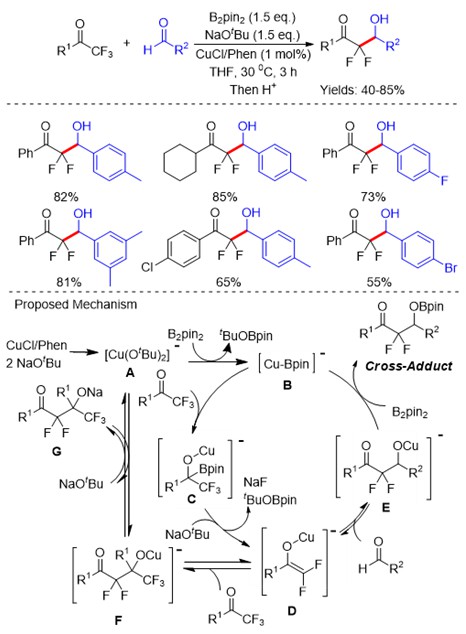
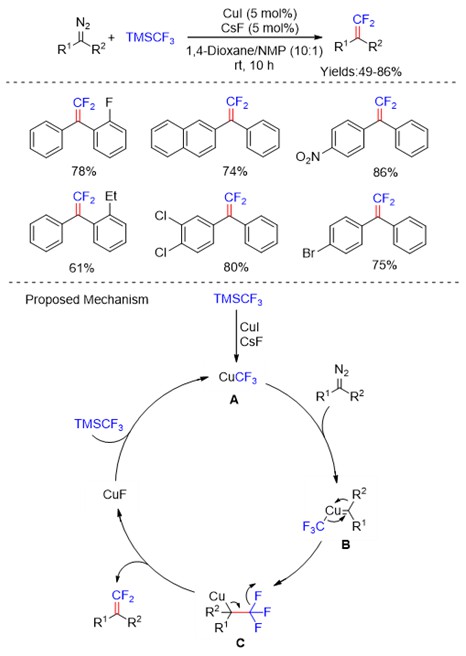
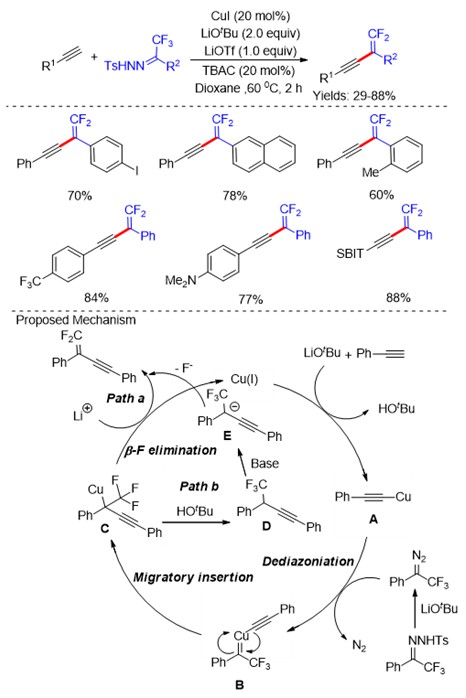
Among small drugs and pharmaceutically relevant molecules, alkaloids have a leading role and those related to pyrrolidine and piperidine are the most common five- and six-membered aliphatic N-heterocycles. Differently from their aromatic counterparts, there are limited strategies to access substituted saturated N-heterocycles with ease and efficiency which fall into two main categories: the cyclization of suitable precursors and the modification of readily available pyrrolidines and piperidines.
Recently, many researchers have dedicated their attention to alternative conditions in order to lower the reaction temperature while keeping the efficiency high in term of both yield and selectivity. In 2010, the group of Köhler observed, for the first time, the acceleration of a Heck reaction under UV-visible light using both homogeneous or heterogeneous Pd(II) pre-catalysts. Few years later, Gevorgyan disclosed an original visible light-induced room temperature Heck reaction between functionalised alkyl halides and styrenes. That was the starting point for the employment of transition metal complexes, especially those containing Pd, as unconventional photocatalysts and remarkable results have been reported by the research groups of Gevorgyan, Fu, Königs, Glorius and Rueping.
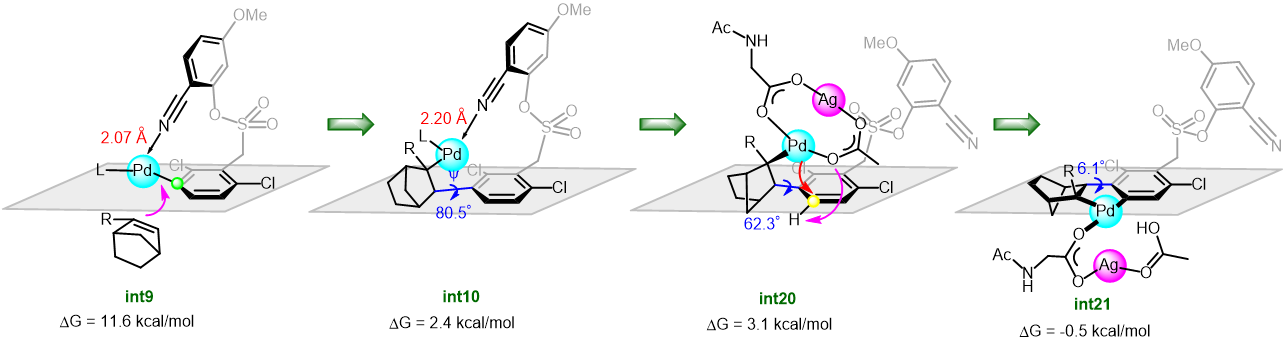
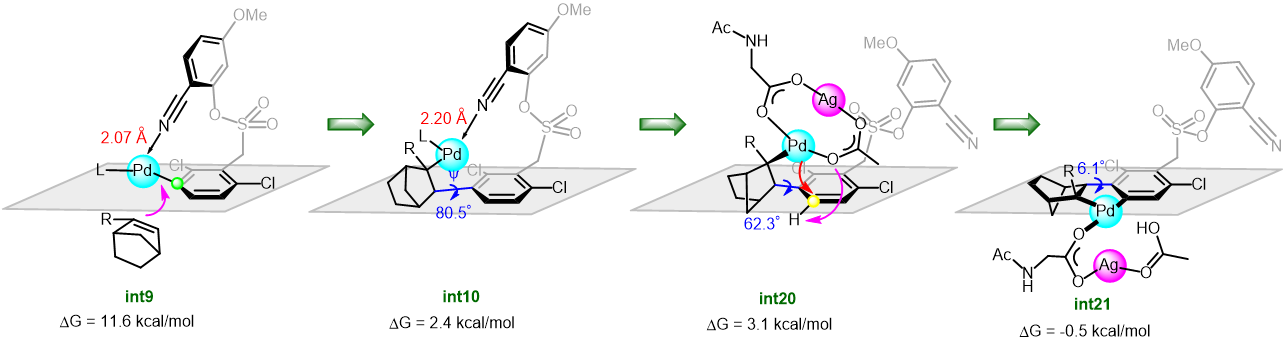
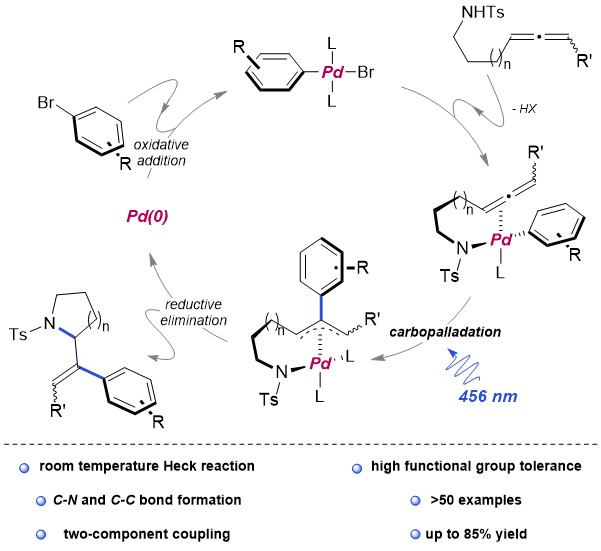
Recently, Renzi et al. presented a synthetic strategy to obtain arylated vinyl pirrolidines and piperidines starting from N-tosylaminoallenes. Key to the process was the usage of the blue light which allowed the efficient exploitation of the simple catalytic system Pd(OAc)2/2PPh3 at room temperature. More than fifty Electron-donating, electron-withdrawing aryl and heteroaryl bromides were coupled with allenes in a Pd(0) catalysed cross coupling. A subsequent domino cyclisation is triggered by the tosylamino functionality. The mechanistic investigation, both experimental and computational, highlighted three main aspects: no radicals seems to be involved in this mechanism, the light is not needed in the oxidative addition of Pd(0) to the aryl bromide because of the associated low energy barrier. On the contrary, the light plays its main role in the carbo-palladation step, but its influence in making easier the formation of the active Pd(0) species cannot be excluded (figure 1).
Corresponding authors:

Annamaria Deagostino
Dept. of Chemistry – University of Torino
Annamaria Deagostino is associate professor of organic chemistry at the University of Torino. She graduated in chemistry at the University of Torino and after a fellowship at the University of Padova, in 1998 she received her Ph.D. in chemistry at the University of Torino, supervisor Prof. Paolo Venturello, and in the same year she obtained a postdoctoral fellowship at the University of Caen, under the supervision of Prof. Marie-Claire Lasne. In 1999, she became assistant professor of organic chemistry at the University of Torino. The main interests of her research group are in the field of synthetic organic chemistry, mainly focused on organopalladium chemistry, visible light photo-catalysis and the synthesis of BNCT (Boron Neutron Capture Therapy) theranostic agents.

Giovanni Ghigo
Dept. of Chemistry – University of Torino
Giovanni Ghigo is assistant professor of organic chemistry at the University of Torino since 2005. He graduated in chemistry 1994 and in 1999 received his Ph.D. in Chemistry at the University of Torino, supervisor Prof. Glauco Tonachini. He obtained postdoctoral fellowships at the University of Torino then at the University of Lund.
GG is a computational organic chemist interested in the study of the mechanisms of a large type of organic reactions spanning from the generation and oxidation of organic pollutants and, more recently, metal-free, copper or palladium catalyzed and photocatalyzed reactions with synthetic applications.








")