Disulfide bond reduction-triggered molecular hydrogels of folic acid–Taxol conjugates
Chengbiao Yang, Dongxia Li, Qianqi FengZhao, Lianyong Wang, Ling Wang and Zhimou Yang
Org. Biomol. Chem., 2013, DOI: 10.1039/C3OB40969D, Paper

The influence of G-quadruplex structure on DNA-based asymmetric catalysis using the G-quadruplex-bound cationic porphyrin TMPyP4·Cu
Michael Wilking and Ulrich Hennecke
Org. Biomol. Chem., 2013, DOI: 10.1039/C3OB41366G, Paper

A highly active cyclometallated iridium catalyst for the hydrogenation of imines
Barbara Villa-Marcos, Weijun Tang, Xiaofeng Wu and Jianliang Xiao
Org. Biomol. Chem., 2013, DOI: 10.1039/C3OB41150H, Paper

ω-Transaminase-catalyzed asymmetric synthesis of unnatural amino acids using isopropylamine as an amino donor
Eul-Soo Park, Joo-Young Dong and Jong-Shik Shin
Org. Biomol. Chem., 2013, DOI: 10.1039/C3OB40495A, Paper

Visible-light activatable organic CO-releasing molecules (PhotoCORMs) that simultaneously generate fluorophores
Ping Peng, Chaoming Wang, Zheng Shi, Valentine K. Johns, Liyuan Ma, Jeremiah Oyer, Alicja Copik, Robert Igarashi and Yi Liao
Org. Biomol. Chem., 2013, DOI: 10.1039/C3OB41385C, Communication

The first asymmetric total synthesis of (+)-coriandrone A and B
Wenjing Wang, Jijun Xue, Tian Tian, Yingdong Jiao and Ying Li
Org. Biomol. Chem., 2013, DOI: 10.1039/C3OB41497C, Communication
-coriandrone A and B")
Convergent synthesis and cellular uptake of multivalent cell penetrating peptides derived from Tat, Antp, pVEC, TP10 and SAP
Gabriela A. Eggimann, Stefanie Buschor, Tamis Darbre and Jean-Louis Reymond
Org. Biomol. Chem., 2013, DOI: 10.1039/C3OB41023D, Paper

Copper(II) chloride mediated (aza)oxindole synthesis by oxidative coupling of Csp2–H and Csp3–H centers: substrate scope and DFT study
Chandan Dey, Evgeny Larionov and E. Peter Kündig
Org. Biomol. Chem., 2013, DOI: 10.1039/C3OB41254G, Paper
 chloride mediated (aza)oxindole synthesis by oxidative coupling of Csp2–H and Csp3–H centers")
Histidine-functionalized water-soluble nanoparticles for biomimetic nucleophilic/general-base catalysis under acidic conditions
Geetika Chadha and Yan Zhao
Org. Biomol. Chem., 2013, DOI: 10.1039/C3OB41485J, Paper

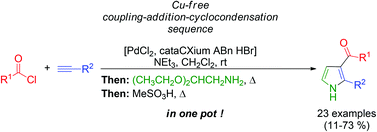
A one-pot coupling–addition–cyclocondensation sequence (CACS) to 2-substituted 3-acylpyrroles initiated by a copper-free alkynylation
Jan Nordmann and Thomas J. J. Müller
Org. Biomol. Chem., 2013, DOI: 10.1039/C3OB41269E, Paper








")
















-complexes of glycine")





{kind=link}