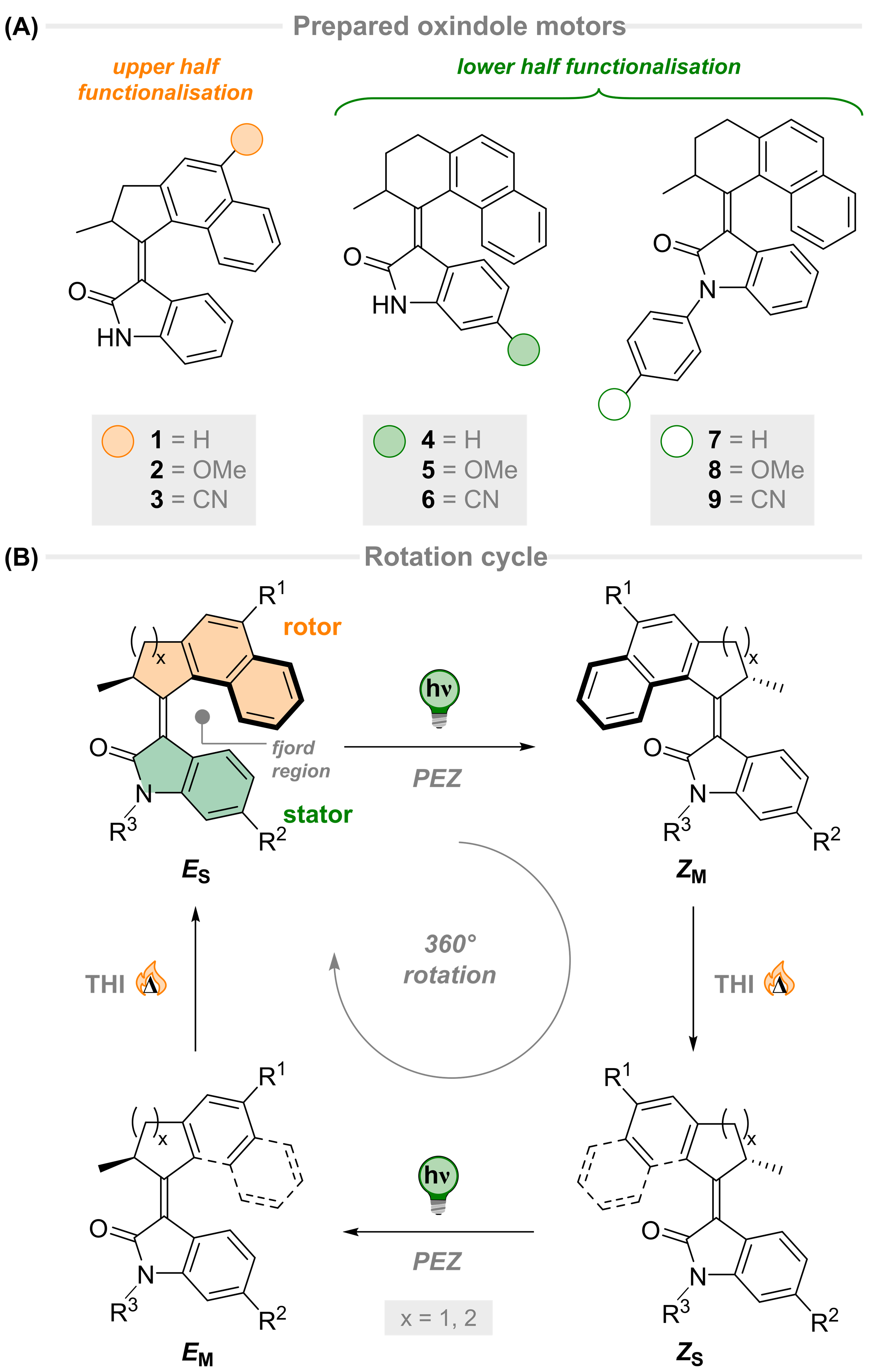
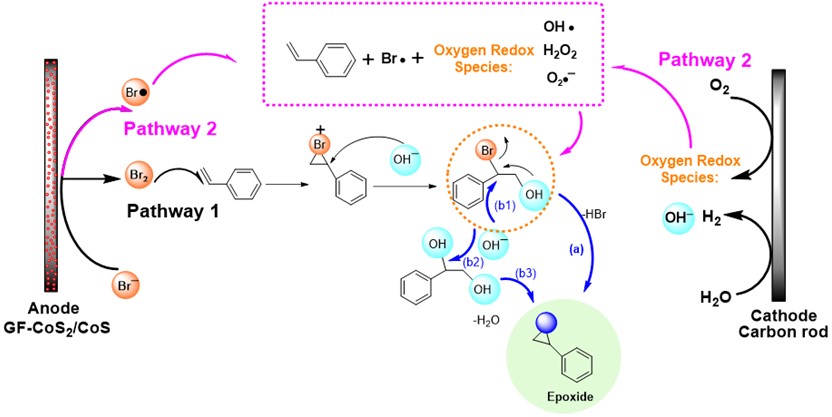
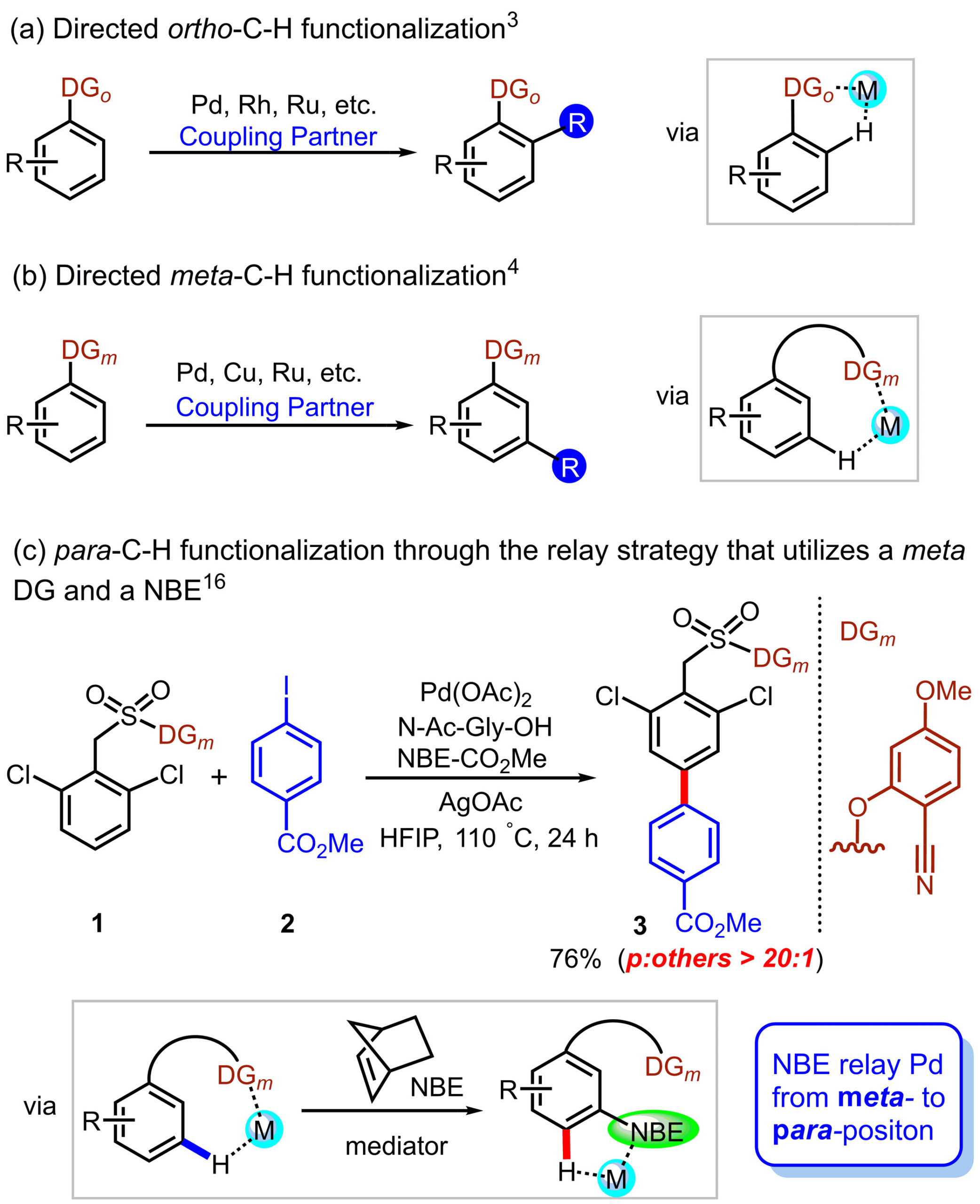
Carbon–phosphorus (C–P) bond cleavage reactions are one of the most important transformations because organophosphorus compounds are widely utilized as pharmaceuticals, phosphine ligands, organocatalysts, and functional materials. Therefore, C–P bond cleavage reactions have been developed using various approaches such as radicals produced using photolysis or peroxides, reduction using transition metal catalysts and alkali metals, and via P(V) intermediates (Scheme 1A). However, these synthetic methodologies require relatively harsh conditions, leading to low functional group tolerance. In addition, they require the use of precious transition metals and the formation of highly polarized C–P bonds in activated species.
Recently, the group of Prof. Shinobu Takizawa and Mohamed S. H. Salem as a collaborator from SANKEN, Osaka University have developed a metal-free C(aryl)–P bond cleavage reaction starting via the reaction of triarylphosphines and alkynyl esters using water as a nucleophile and electrophile to yield propanoic acid/phosphine oxide derivatives (Scheme 1B), which have applications in pharmaceuticals and catalysis.
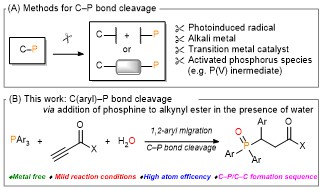
Scheme 1: Methods of C(aryl)–P bond cleavage.
The reaction proceeded under mild and neutral conditions (10 °C, 24 h, H2O as a nucleophile) without using any metal catalyst, which is consistent with the concepts of green chemistry. In addition, they have carried out the full optimization of the reaction system to obtain high yields (up to 98%). The scopes of phosphine reagents and activated alkynes were investigated under the optimal conditions to disclose a broad spectrum with high yields in most of the cases.
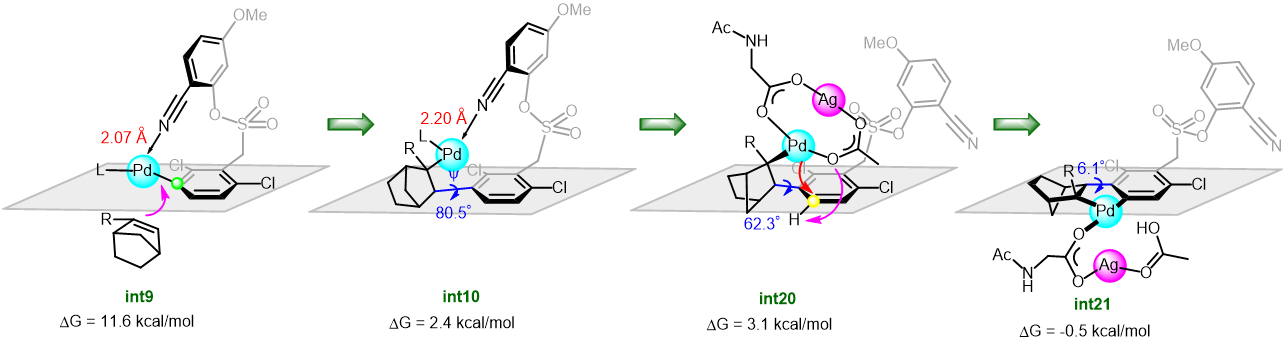
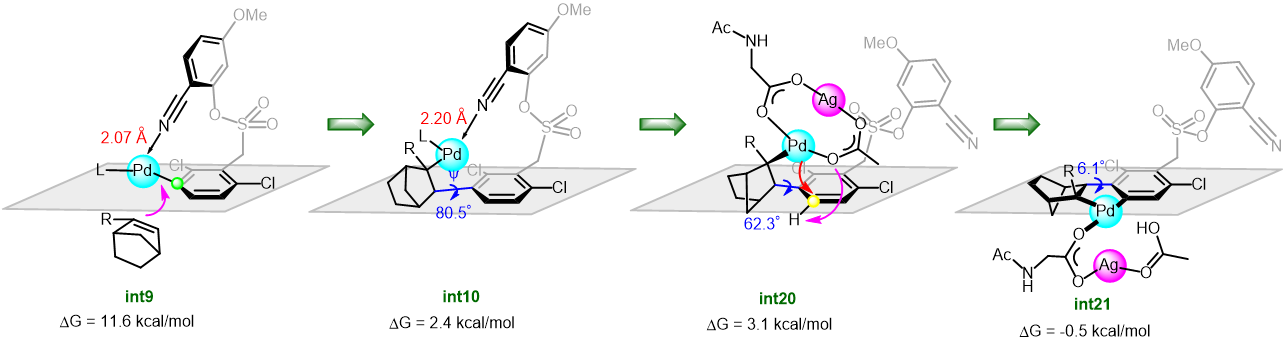
Takizawa, Salem and coworkers have complemented their experimental work with computational calculations to understand and verify their proposed reaction mechanism. DFT calculations and some control experiments using (allenic esters, or vinyl ketone instead of propiolate esters), and (D2O, or H218O instead of water) indicated that the rapid formation of hydroxy-λ5-phosphane as a key intermediate plays a crucial role in smooth C(aryl)–P bond cleavage (Scheme 2).
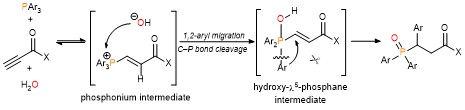
Scheme 2: The proposed reaction mechanism of rapid C(aryl)–P bond cleavage and 1,2-aryl migration to the Michael acceptor via a hydroxyl-λ5-phosphane intermediate.
The development of this new synthetic method using metal-free conditions is crucial for the development of greener chemical practices. Further, the understanding of the mechanism of these synthetic reactions has a key value in expanding their applications, as well as the synthesis of industrially important phosphorus-containing molecules.
Author:

Mohamed S. H. SALEM
Osaka University
Mohamed Salem is a Ph.D. student in the SANKEN at Osaka University (Japan). He received his M.Sc. in Medicinal Chemistry at the Faculty of Pharmacy, Suez Canal University in 2017. Subsequently, he received a Japanese Government Scholarship (MEXT) to study for a Ph.D. in 2019 with Professor Hiroaki SASAI and Shinobu TAKIZAWA. The research field of Mohamed Salem is asymmetric catalysis using first-row organometallic complexes such as Cobalt and Vanadium and the study and development of new green and cutting edge electrochemical transformations.
https://orcid.org/0000-0002-8919-095X
https://www.researchgate.net/profile/Mohamed-Salem-39
Corresponding Author:

Shinobu TAKIZAWA 滝澤 忍
Osaka University
Shinobu TAKIZAWA is an associate professor in the Institute of Scientific and Industrial Research (SANKEN) and AI Research Center at Osaka University (Japan). He received his Ph.D. in chemistry at the Graduate School of Pharmaceutical Science, Osaka University in 2000. From 2006 to 2008 he did postdoctoral studies at The Scripps Research Institute (USA) with Professor Dale L. Boger. The research field of Professor Shinobu TAKIZAWA is asymmetric catalysis using either organo-or organometallic catalysts and the study and development of new green and cutting edge chemical transformations either electrochemically or photochemically guided by AI and ML. He is the author of more than 118 articles indexed by SCI and cited more than 4000 times with an index H = 38.
https://orcid.org/0000-0002-9668-1888








")