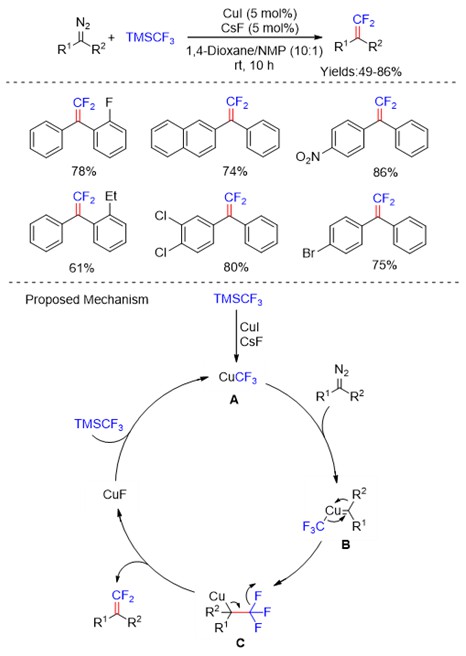
Recent advances in chemistry, synthetic biology, and material science have enabled the development of cell membrane-based drug delivery systems (DDSs), often referred to as “artificial cells” or protocells. In particular, the use of these cellular mimics for directed delivery and controlled release of therapies has burgeoned with advances in molecular biology, proteomics, nanotechnology, biotechnology, and polymer chemistry. Our paper focuses on a discussion on the concept of building simple functional units that recapitulate living cells, protocells, that can respond to and deliver targeted therapies. Artificial cells can be made by removing functions from natural systems in a top-down manner, or assembly from synthetic, organic, or inorganic materials, through a bottom-up approach where simple units are integrated to form more complex structures.
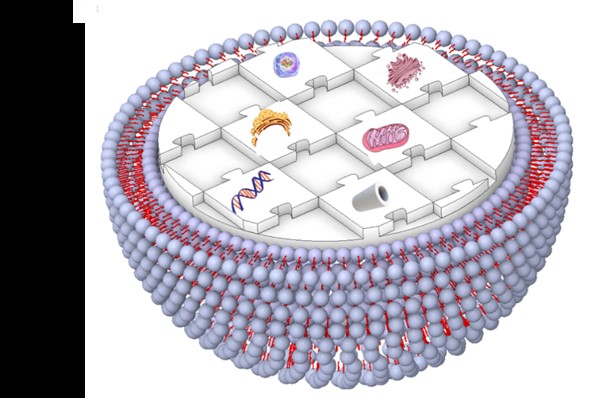
Fig. 1 Schematic illustration representing a human-made artificial cell comprising of nucleic acids, cytoskeleton, small biomolecules, and cytoplasmic organelles in a giant unilamellar vesicle.
Artificial cells that use liposomes, polymersomes, or dendrimersomes to create a membrane component have been developed as advanced DDSs for cancer treatments, gene therapies, and vaccines. Surface modification of these carriers can improve payload delivery, through increasing circulation times and preventing degradation, and enable systems with active-targeting and stimulus-responsive capabilities. Overall, artificial cells can be fabricated with various sizes, deformability, sustained/controlled payload release profiles, and versatile targeting functions. There are still remarkable differences between the living cellular membranes and their artificial compartments, and studies to develop new materials as well as basic experiments to better understand how a membrane’s composition contributes to its physical properties and biological functions are important to bridge this gap. Therefore, the development of alternative and new smart and improved properties such as nanoscale efficiency, self-organization, and adaptability for therapeutic and diagnostic applications is needed. Ultimately, creating artificial cells with properties and functions of living cells and have broad clinical utility will require extensive cross-disciplinary cooperation among the fields of biophysics, biochemistry, medicine, biomedical engineering, materials science, and molecular/cellular biology.

Corresponding author: Nureddin Ashammakhi
Nureddin Ashammakhi is focusing on translational tissue regenerative therapy. Currently, he is working on 3D bioprinting and organ-on-a-chip models for regenerative and personalized medicine. He is an expert in bioabsorbable, nanofibrous, and drug release implants. He was previously a professor of biomaterials technology in the Tampere University of Technology, Finland, Chair of regenerative medicine, Keele University, UK, and Adjunct Professor in Oulu University, Finland before he joined UCLA as a visiting professor, then as Associate Director of the Center for Minimally Invasive Therapeutics and Adjunct Professor after which he joined Michigan State University.
Contact information:
Department of Bioengineering, University of California, Los Angeles, 420 Westwood Plaza, Engineering V, Los Angeles CA 90095, USA, Email: n.ashammakhi@ucla.edu, n.ashammakhi@gmail.com








")