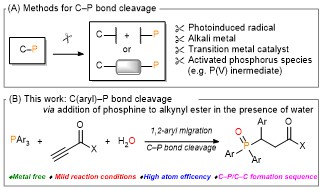
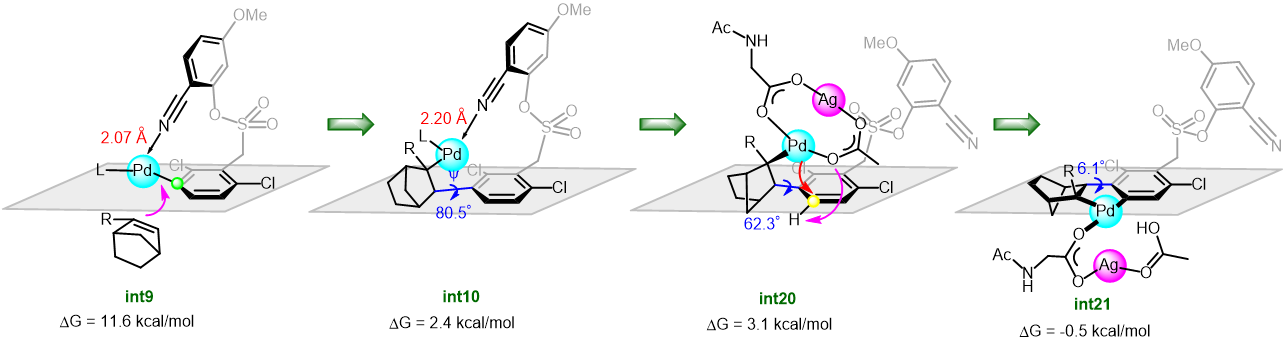
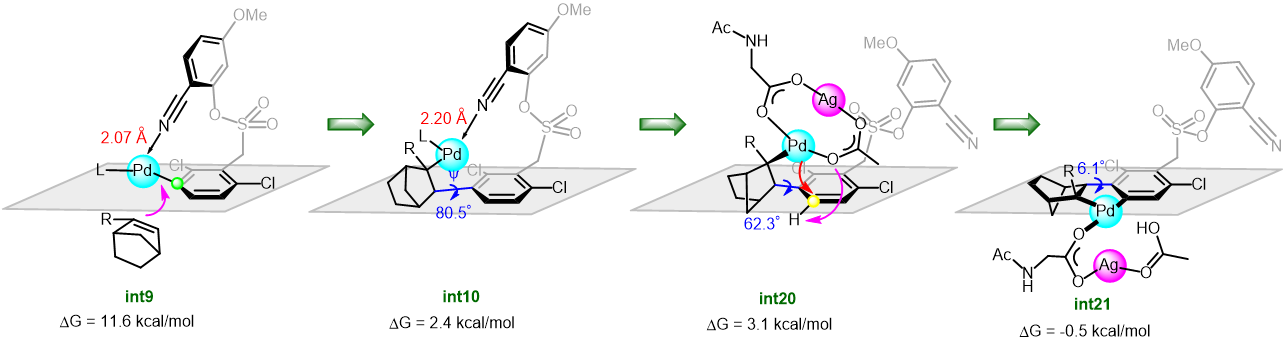
Cycloisomerization of 1,n-enynes provides a variety of cyclic compounds via multiple bond cleavage and formation. Since these processes are generally induced by alkyne π-bond activation, transition metal catalysts have been well studied for the selective construction of these cyclic products (Scheme 1A). However, transition metal catalysts can be hardly applied to electron-deficient alkynes having acyl groups or other relative strong electron-withdrawing groups. Since acyl groups can be converted to various functional groups, it is desirable to develop catalyst systems applicable to such starting materials. In addition, the activation of alkynes via carbonyl groups can lead to the construction of new skeletons and thus systematic studies on the cycloisomerization of enynones is an important research project.
Recently, as a further expansion of BF3·MeCN-catalyzed cycloisomerization of 7-en-2-ynones into six-membered cyclic dienes (Org. Lett., 2020, 22, 4063), the group of Prof. Akio Saito from Tokyo University of Agriculture and Technology have developed the aluminum halide-mediated formation of the halogenated bicyclo[3.1.0]hexanes from the same substrates (Scheme 1B).
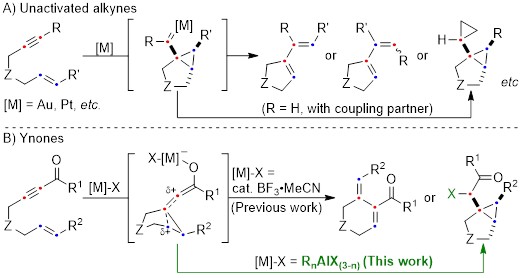
Scheme 1: Cycloisomerization of enynes and enynones.
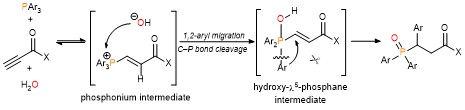
The aluminum halide-mediated cycloisomerization of 7-en-2-ynones proceeded at room temperature to give the corresponding the halogenated bicyclo[3.1.0]hexanes in 45-96% yields. Furthermore, quenching with NCS (N-chlorosuccinimide) before the usual work-up afforded the dichloroketone product in good yield through the chlorination of the aluminum enolate intermediate (Scheme 2).
On the basis of DFT calculations and experimental data, we proposed the mechanism including the common zwitterionic intermediate with six-membered cyclic dienes (Scheme 1B), and concluded that different bond lengths between group 13 elements and halogens lead to cycloisomerization into different products.
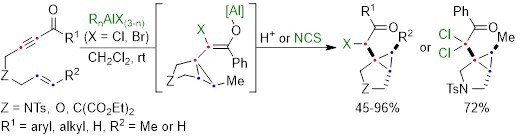
Scheme 2: Aluminum halide-mediated cycloisomerization of 7-en-2-ynones into the halogenated bicyclo[3.1.0]hexanes
Corresponding author:

Akio SAITO Professor
Akio SAITO received his Ph.D. degrees from Tokyo University of Pharmacy and Life Sciences in 2003. From 2001 to 2005, he joined as a Research Associate in the group of Professor Takeo Taguchi at the same university. From 2005 to 2012, he worked as an Assistant Professor with Professor Yuji Hanzawa at Showa Pharmaceutical University. In 2012, he moved to the present university as an Associate Professor. Since 2021, he was appointed to the present position. His current research interests include the development of domino reactions and multicomponent reactions based on iodine and other non-metallic elements chemistry. He is the author of more than 96 articles indexed by WoS and cited more than 2400 times with an h-index = 31.
https://www.webofscience.com/wos/author/record/C-1270-2013








")