Single-molecule magnets (SMMs) are molecules that retain the slow relaxation of their magnetization upon removal of an applied magnetic field, acting as magnets below a characteristic temperature known as the blocking temperature. Research in this field has recently focused on the use of radicals as ligands to enhance the performance of SMMs through the exchange coupling of the radical- and metal-based spins. The strong interaction between the spin of the radical with the unpaired electrons of a metal ion effectively suppresses the quantum tunneling of the magnetization (QTM) and fast spin relaxation pathways involving spin excited states, thus promoting a thermal relaxation pathway for magnetization reversal. The latter is particularly important when designing lanthanide (Ln) based SMMs as the core-like nature of the 4f orbitals makes magnetic coupling challenging and thus they tend to suffer from through-barrier relaxation of the magnetization (i.e., QTM, Raman and direct mechanisms).
To this day several attempts towards this direction have been made leading to the isolation of strongly coupled Ln SMMs with the N2•3--based family exhibiting very good magnetic performance. However, the rational incorporation of the N2 species into complexes is a synthetic challenge and it does not offer any room for structural modification. For this reason, other radicals, such as tetrazines have been explored by the group of Prof. Muralee Murugesu of the University of Ottawa. In the past the researchers had successfully incorporated the 1,2,4,5-tetrazine radical anion (tz•−) into tetranuclear “Ln4” metallocenes which led to strong magnetic coupling and significant magnetic hysteresis (Hc = 3 T).
Recently, the researchers aimed to isolate a dinuclear building block so that the role of the bridging ligand in the overall magnetic coupling in Ln systems can be better understood. In further detail, they have utilized the high performing {Cp*2LnIII}+ moieties and bridge them by employing the tz•−, leading to the isolation of a new family of radical-bridged Ln metallocenes: [(Cp*2LnIII)2(tz•−)(THF)2](BPh4), (Ln = Gd (1), Tb (2), Dy (3); THF = tetrahydrofuran).
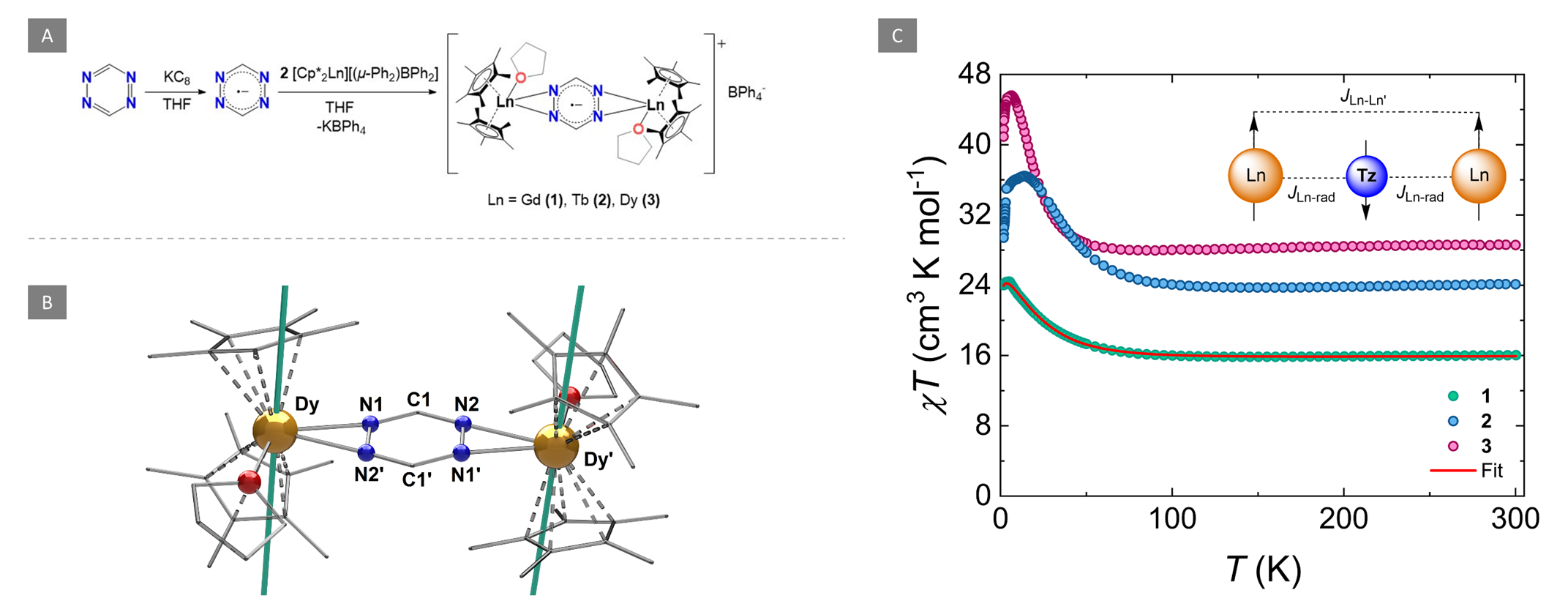
Figure 1. A) Synthesis of the radical-bridged dinuclear compounds (1-3). (B) Molecular structure of 3. Partial labelling and omission of the BPh4- moiety and H-atoms have been employed for clarity. The solid teal lines represent the orientation of the principal magnetic axes of the ground Kramers doublet. C) Variable temperature dc susceptibility of 1 (teal circles), 2 (blue circles) and 3 (magenta circles) under an applied field of 1000 Oe. The solid red line represents the fit as determined from the application of the -2J formalism. Insert: Simplified illustration of the two J-model which was used to fit the data highlighting the antiparallel spin alignment of the LnIII ions with respect to the tz•- ligand.
They showed that a strong magnetic coupling between the LnIII ions and the tz•− was achieved, revealing a JGd-rad = -7.2 cm-1 for 1 which is even comparable to some N2•3- bridged SMMs. Due to this, both 2 and 3 displayed zero-field SMM behavior with slow relaxation of the magnetization and magnetic hysteresis.
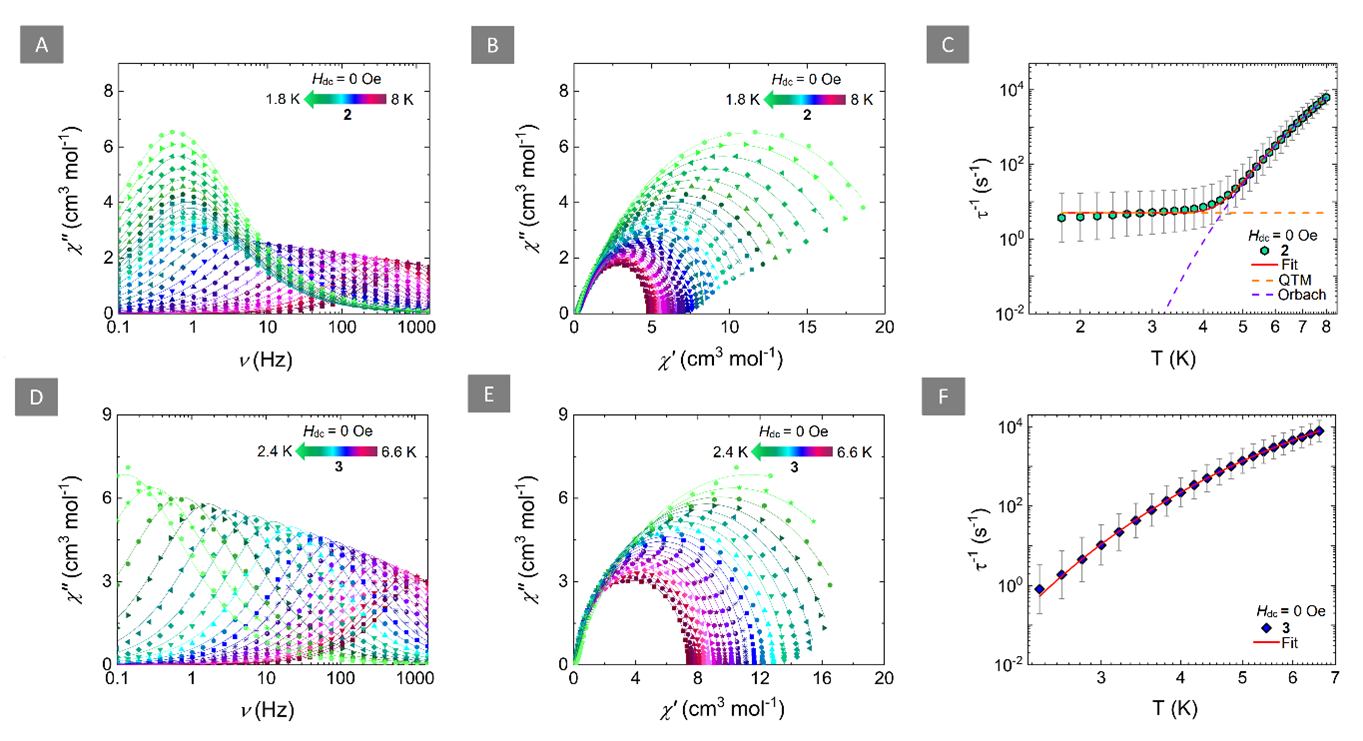
Figure 2 Left: Frequency-dependence of the out-of-phase magnetic susceptibility (χ’’) at zero-field for 2 (A) and 3 (D) at the respective temperature regions. Solid lines represent fits to the generalized Debye model. Middle: Cole-Cole plots for 2 (B) and 3 (E) at the respective temperature regions (Hdc = 0 Oe). Solid lines represent fits to the generalized Debye model. Right: Temperature-dependence of the relaxation times (τ) for 2 (C) and 3 (F) with the respective estimated standard deviations (gray bars). The estimated standard deviations of the τ were calculated from the α-parameters of the generalized Debye fits and the log-normal distribution. The solid red lines represent the best-fit while the dashed orange and purple lines in (C) represent the individual components of the magnetic relaxation for QTM and Orbach processes, respectively.
Ab initio calculations verified the strong antiferromagnetic Ln-rad coupling and showed that the magnetic state of 2 and 3 can be interpreted as a “giant-spin” where the relaxation of the magnetization is related to changes in the magnetic state of the overall exchange-coupled system. The slow relaxation of these SMMs is mediated via thermally activated processes through the first excited KDs which correspond to an Ising-type ferrimagnetic spin configuration where the magnetic moments of the LnIII ions are co-aligned while the magnetic moment of the radical points to the opposite direction.
The researchers believe that the results presented in this work will be helpful for future strategies on designing new lanthanide metal complexes employing radical ligands in the pursuit of new strongly-coupled zero field SMMs.
Corresponding author:

Prof. Muralee Murugesu
University of Ottawa
Prof. Muralee Murugesu received his PhD from the University of Karlsruhe in 2002 under the supervision of Prof. A. K. Powell. He undertook postdoctoral stays at the University of Florida (2003–2005) with Prof. G. Christou, and jointly at the University of California, Berkeley and the University of California, San Francisco under the supervision of Prof. J. R. Long and the Nobel Laureate Prof. S. Pruissner (2005–2006). In 2006, he joined the University of Ottawa as an assistant professor and since 2015 he is a full professor. Prof. Murugesu’s research focuses on the design and development of Single-Molecule Magnets, Metal-Organic Frameworks and High-Energy materials.








")