Since Wilson and Flanigen found the first microporous aluminophosphate (AlPO) in 1982, the synthesis of AlPO materials with novel frameworks and study their potential properties, such as adsorption, catalysis, and separation, has attracted intense interest of researchers. As the main family of AlPO based materials, 2D layered materials show a rich variety of structures and compositions depending on the diverse coordination and connection modes of Al and P atoms. In recent years, 2D materials, possessing highly exposed surfaces and diverse structures, have become a dramatic category of supports. However, 2D molecular sieves are serving as catalyst supports are very few.
Recently, Prof. Jiuxing Jiang at Sun Yat-sen University and Dr. Jiang-Zhen Qiu of Zhongkai University of Agriculture and Engineering synthesized a new 2D layered aluminophosphate compound by adopting rigid and bulky template of 3,5,N,N-tetramethyladamantane-1-amine (ada) through hydrothermal conditions, which is named the Zhongkai University of Agriculture and Engineering NO.1 (ZHKU-1), with the composition |Hada|6[Al6(PO4)8](H2O)11. The structure of ZHKU-1 was constructed from the alternate connection of AlO4 and triply bridged PO4 tetrahedra ([O=PO3]3-) to form a 4, 6, 12-net (Figure 1). The inorganic sheets are linked and separated by protonated amine and H2O molecules by extensive H-bonds, giving a new 2D structure with an interlayer space of 19.6 Å.
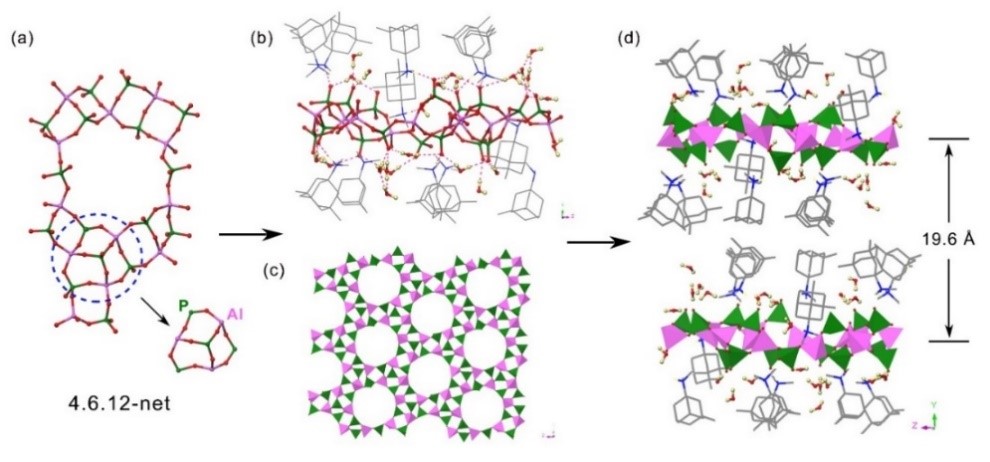
Figure 1 Crystal structure figures of ZHKU-1
ZHKU-1, as a 2D material with a more exposed surface, could be adapted for encapsulating metal nanoparticles (NPs). Ag species are immobilized on the supports of ZHKU-1 by UV reduction and deposition, forming the catalyst of Ag@ZHKU-1 with high loading of 4.9 wt%. The HRTEM images reveal the highly uniform Ag clusters with visually observed sizes of 1.9 nm (Figure 2).
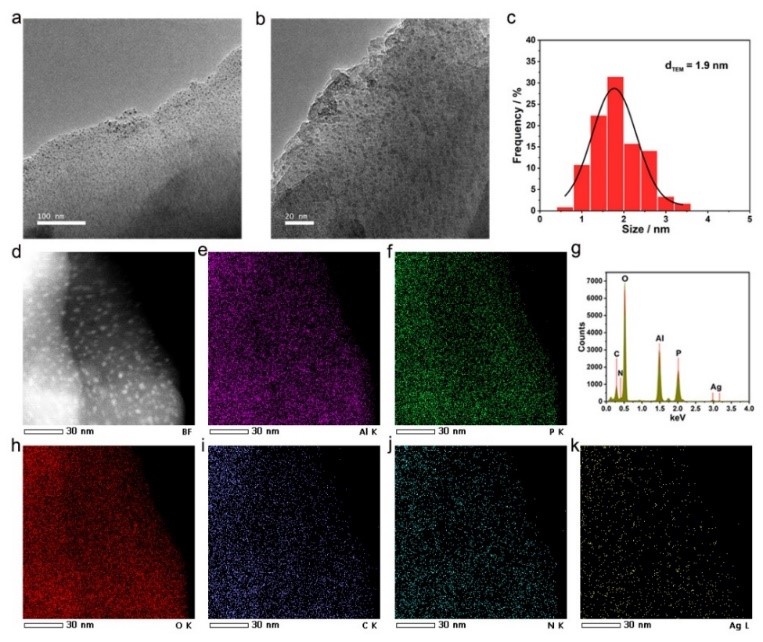
Figure 2 The characterizations of Ag@ZHKU-1 for HRTEM and corresponding mapping images
Since 4-nitrophenol (4-NP) is a notorious industrial pollutant, Ag@ZHKU-1 is applied to the model reaction for 4-NP reduction. The catalytic results show that the reduction reaction of 4-NP into 4-aminophenol could be completely performed within 75 s in NaBH4 solution (Figure 3). Moreover, the catalytic activity was almost the same, with almost 99% conversion after eight cycles. The remarkable catalytic activity and recyclability of Ag@ZHKU-1can be attributed to the high dispensability of Ag nanoclusters confined to the 2D support, which provides more accessible Ag active sites to 4-NP.
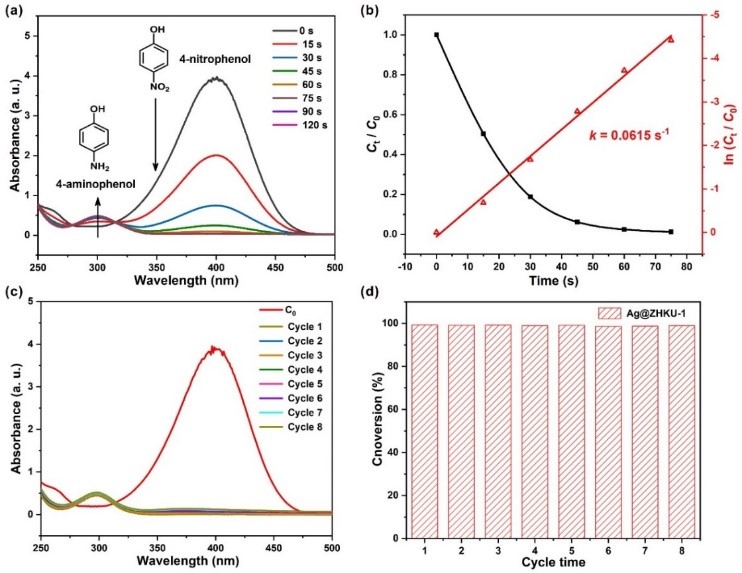
Figure 3 Catalytic activity and recycling tests for 4-nitrophenol reduction are over Ag@ZHKU-1 catalyst
This work reports a new 2D layered aluminophosphate compound, which expends the aluminophosphates family. Furthermore, the confinement of metal Ag on this new layer structure through photodeposition gives rise to a small size (~1.9 nm) of AgNPs with homogeneous dispersion. The catalyst of Ag@ZHKU-1 shows excellent catalytic activity and high conversion for 4-nitrophenol reduction. This work is significant for designing 2D aluminophosphate materials to confine small metal nanoparticles for catalytic application.

Prof: Jiuxing Jiang (ORCID: 0000-0001-9664-3235). I received Ph.D degree from Jilin University in 2010. Afterward, I spent 5 years for a Post Doc. stay in Instituto Technologia Quimica (UPV-CSIC) in Valencia Spain supervised by Prof: Avelino Corma. After independent work in Sun Yat-sen University (2015-now), I keep my interesting on the topologically new zeolite synthesis (four three letter code, IRR, -IRY, -IFU, -SYT were granted by Structure Committee of Internation Zeolite Association), and zeolite based heterogeneous catalysis, such as: acid-base catalysis, catalytic ammonium synthesis, NH3-SCR for deNOx reaction, porous materials for energy storage, etc. I have published more than 20 high-impact journal articles, such as Science, Angew. Chem. Int, Ed, Chem.Sci, Chem. Mater. etc. Among them, the work on the synthesis and structure of zeolite ITQ-43 was published in Science, 2011, 333, 1131-1134 and was selected as annually breakthrough of 2011 by Science. Currently serve as a member of Zeolite Committee of Chinese Chemical Society and youth editorial member of Journal of Chemical Research in Chinese University.

Jiang-Zhen Qiu: She received her Ph.D. degree in 2019 from Sun Yat-sen University with M.S. Supervisor Prof. Ming-Liang Tong and Ph.D. Supervisor Prof. Jiuxing Jiang. In 2020, she was introduced to Chemical Engineering of Zhongkai University of Agriculture and Engineering as an “Excellent Doctor”. Her interested research fields include synthesizing new topological structures of zeolite and exploring multifunctional materials with novel functionalities such as light, conductivity, magnetism, or catalysis. Currently, she and her collaborators published 10 papers in related fields, such as Chem. Mater., Chem. Sci., Inorg. Chem. Front., Chem. Commun., Chem. Eur. J.










")