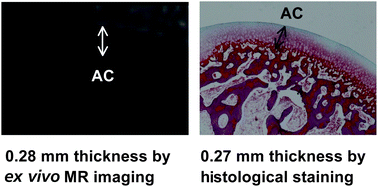
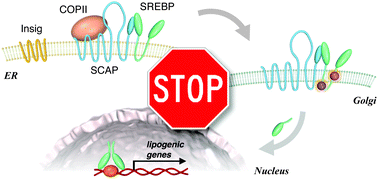
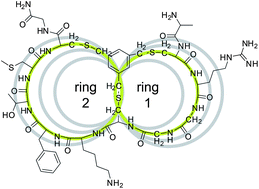
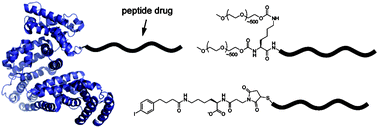
| A recent workshop on the role of computer aided drug discovery was jointly held by the Royal Society of Chemistry and the Biochemical Society with the aim of aiding collaborations between biological scientists and computational chemists.
Following this workshop we have brought together a collection of articles and books that highlight recent research on computer-aided drug discovery and related areas. Below is a slection from across the Royal Society of Chemistry and you can also find a collection of articles from the Biochemical Society HERE…
All of the Communications, Papers and Reviews below are free to access until 7 February 2014.
|
Books
(PDFs of the front matter, table of contents and first chapter are free to view.)
 Physico-Chemical and Computational Approaches to Drug Discovery
Physico-Chemical and Computational Approaches to Drug Discovery
Editors: Javier Luque, Xavier Barril
Drug Design Strategies
Editors: David J Livingstone, Andrew M Davis
Innovations in Biomolecular Modeling and Simulations: Volume 2:
Chapter 11 – Structure-based Design Technology CONTOUR and its Application to Drug Discovery
Zhijie Liu, Peter Lindblom, David A. Claremon and Suresh B. Singh
Chapter 12 – Molecular Simulation in Computer-aided Drug Design: Algorithms and Applications
Robert V. Swift and Rommie E. Amaro
Chapter 13– Computer-aided Drug Discovery: Two Antiviral Drugs for HIV/AIDS
J. Andrew McCammon
G Protein-Coupled Receptors: From Structure to Function:
Chapter 18 – Structure-based Virtual Screening for Ligands of G Protein-coupled Receptors
Stefano Costanzi
Reviews
Informatic strategies for the discovery of polyketides and nonribosomal peptides
Chad Johnston, Ashraf Ibrahim and Nathan Magarvey
Med. Chem. Commun., 2012, DOI: 10.1039/C2MD20120H, Review Article
Molecular docking for virtual screening of natural product databases
Dik-Lung Ma, Daniel Shiu-Hin Chan and Chung-Hang Leung
Chem. Sci., 2011, DOI: 10.1039/C1SC00152C, Minireview
Approaches to discover non-ATP site kinase inhibitors
Lori Krim Gavrin and Eddine Saiah
Med. Chem. Commun., 2013, DOI: 10.1039/C2MD20180A, Review Article
Drug repositioning by structure-based virtual screening
Dik-Lung Ma, Daniel Shiu-Hin Chan and Chung-Hang Leung
Chem. Soc. Rev., 2013, DOI: 10.1039/C2CS35357A, Review Article
Selective inhibition of the unfolded protein response: targeting catalytic sites for Schiff base modification
Susana M. Tomasio, Heather P. Harding, David Ron, Benedict C. S. Cross and Peter J. Bond
Mol. BioSyst., 2013, DOI: 10.1039/C3MB70234K, Review Article
Network-based drug repositioning
Zikai Wu, Yong Wang and Luonan Chen
Mol. BioSyst., 2013, DOI: 10.1039/C3MB25382A, Review Article
Communications and Papers
Virtual screening and experimental validation reveal novel small-molecule inhibitors of 14-3-3 protein–protein interactions
Philipp Thiel, Lars Röglin, Nicole Meissner, Sven Hennig, Oliver Kohlbacher and Christian Ottmann
Chem. Commun., 2013, DOI: 10.1039/C3CC44612C, Communication
Plugging the explicit σ-holes in molecular docking
Michal Kolář, Pavel Hobza and Agnieszka K. Bronowska
Chem. Commun., 2013, DOI: 10.1039/C2CC37584B, Communication
Fragments to link. A multiple docking strategy for second site binders
Márton Vass and György M. Keserű
Med. Chem. Commun., 2013, DOI: 10.1039/C2MD20267K, Concise Article
A rapid identification of hit molecules for target proteins via physico-chemical descriptors
Goutam Mukherjee and B. Jayaram
Phys. Chem. Chem. Phys., 2013, DOI: 10.1039/C3CP44697B, Paper
 Discovery of novel inhibitors for human farnesyltransferase (hFTase) via structure-based virtual screening
Discovery of novel inhibitors for human farnesyltransferase (hFTase) via structure-based virtual screening
Xiaojuan Yu, Xue Zhao, Lili Zhu, Chuanxin Zou, Xiaofeng Liu, Zhenjiang Zhao, Jin Huang and Honglin Li
Med. Chem. Commun., 2013,DOI: 10.1039/C3MD00058C, Concise Article
Prediction of chemical–protein interactions: multitarget-QSAR versus computational chemogenomic methods
Feixiong Cheng, Yadi Zhou, Jie Li, Weihua Li, Guixia Liu and Yun Tang
Mol. BioSyst., 2012, DOI: 10.1039/C2MB25110H, Paper
Identification of novel inhibitors of p53–MDM2 interaction facilitated by pharmacophore-based virtual screening combining molecular docking strategy
Weisi Wang, Xiaolei Zhu, Xueqin Hong, Lin Zheng, Hong Zhu and Yongzhou Hu
Med. Chem. Commun., 2013, DOI: 10.1039/C2MD20208E, Concise Article
Predicting cancer drug mechanisms of action using molecular network signatures
Justin R. Pritchard, Peter M. Bruno, Michael T. Hemann and Douglas A. Lauffenburger
Mol. BioSyst., 2013, DOI: 10.1039/C2MB25459J, Paper
Prospective use of molecular field points in ligand-based virtual screening: efficient identification of new reversible Cdc25 inhibitors
James C. Collins, Alan Armstrong, Kathryn L. Chapman, Hayley C. Cordingley, Albert A. Jaxa-Chamiec, Katie E. Judd, David J. Mann, Katherine A. Scott, Catherine J. Tralau-Stewart and Caroline M. R. Low
Med. Chem. Commun., 2013, DOI: 10.1039/C3MD00047H, Concise Article
Discovery of Rho-kinase inhibitors by docking-based virtual screening
Mingyun Shen, Huidong Yu, Youyong Li, Pixu Li, Peichen Pan, Shunye Zhou, Liling Zhang, Shang Li, Simon Ming-Yuen Lee and Tingjun Hou
Mol. BioSyst., 2013, DOI: 10.1039/C3MB00016H, Paper
Prediction of adverse drug reactions by a network based external link prediction method
Jiao Lin, Qifan Kuang, Yizhou Li, Yongqing Zhang, Jing Sun, Zhanling Ding and Menglong Li
Anal. Methods, 2013, DOI: 10.1039/C3AY41290C, Paper
Targeting the inactive conformation of protein kinases: computational screening based on ligand conformation
Pascal Bonnet, Daniel Mucs and Richard A. Bryce
Med. Chem. Commun., 2012, DOI: 10.1039/C1MD00256B, Concise Article
 Structure-oriented bioinformatic approach exploring histidine-rich clusters in proteins
Structure-oriented bioinformatic approach exploring histidine-rich clusters in proteins
Shujian Cun, Yau-Tsz Lai, Yuen-Yan Chang and Hongzhe Sun
Metallomics, 2013, DOI: 10.1039/C3MT00026E, Concise Article
Rational design of small modified peptides as ACE inhibitors
Daniel G. Silva, Matheus P. Freitas, Elaine F. F. da Cunha, Teodorico C. Ramalho and Cleiton A. Nunes
Med. Chem. Commun., 2012, DOI: 10.1039/C2MD20214J, Concise Article
Development of a novel class of B-RafV600E-selective inhibitors through virtual screening and hierarchical hit optimization
Xiangqian Kong, Jie Qin, Zeng Li, Adina Vultur, Linjiang Tong, Enguang Feng, Geena Rajan, Shien Liu, Junyan Lu, Zhongjie Liang, Mingyue Zheng, Weiliang Zhu, Hualiang Jiang, Meenhard Herlyn, Hong Liu, Ronen Marmorstein and Cheng Luo
Org. Biomol. Chem., 2012, DOI: 10.1039/C2OB26081F, Paper
Computational design of a thermostable mutant of cocaine esterase via molecular dynamics simulations
Xiaoqin Huang, Daquan Gao and Chang-Guo Zhan
Org. Biomol. Chem., 2011, DOI: 10.1039/C0OB00972E, Paper
Structure determinants of indolin-2-on-3-spirothiazolidinones as MptpB inhibitors: An in silico study
Yinfeng Yang, Jinghui Wang, Yan Li, Wei Xiao, Zhenzhong Wang, Jingxiao Zhang, Weimin Gao, Shuwei Zhang and Ling Yang
Soft Matter, 2013, DOI: 10.1039/C3SM51995C, Paper
Synthesis of novel PPARα/γ dual agonists as potential drugs for the treatment of the metabolic syndrome and diabetes type II designed using a new de novo design program PROTOBUILD
Yushma Bhurruth-Alcor, Therese Røst, Michael R. Jorgensen, Christos Kontogiorgis, Jon Skorve, Robert G. Cooper, Joseph M. Sheridan, William D. O. Hamilton, Jonathan R. Heal, Rolf K. Berge and Andrew D. Miller
Org. Biomol. Chem., 2011, DOI: 10.1039/C0OB00146E, Paper
Anticancer loading and controlled release of novel water-compatible magnetic nanomaterials as drug delivery agents, coupled to a computational modeling approach
Pierre Dramou, Pengli Zuo, Hua He, Lien Ai Pham-Huy, Wenyue Zou, Deli Xiao, Chuong Pham-Huy and Theophilus Ndorbor
J. Mater. Chem. B, 2013, DOI: 10.1039/C3TB20502A, Paper
An NMR crystallography DFT-D approach to analyse the role of intermolecular hydrogen bonding and π–π interactions in driving cocrystallisation of indomethacin and nicotinamide
Dmytro V. Dudenko, Jonathan R. Yates, Kenneth D. M. Harris and Steven P. Brown
CrystEngComm, 2013, DOI: 10.1039/C3CE41240G, Paper
Modelling the possible bioactivity of ellagitannin-derived metabolites. In silico tools to evaluate their potential xenoestrogenic behavior
Luca Dellafiora, Pedro Mena, Pietro Cozzini, Furio Brighenti and Daniele Del Rio
Food Funct., 2013,4, DOI: 10.1039/C3FO60117J, Paper
Towards ab initio screening of co-crystal formation through lattice energy calculations and crystal structure prediction of nicotinamide, isonicotinamide, picolinamide and paracetamol multi-component crystals
H. C. Stephen Chan, John Kendrick, Marcus A. Neumann and Frank J. J. Leusen
CrystEngComm, 2013, DOI: 10.1039/C3CE40107C, Paper
You may also be interested in the upcoming Faraday Discussion on Molecular Simulations and Visualization in 2014 – Find out more here…
Comments Off on A Focus On Computer-Aided Drug Discovery








")