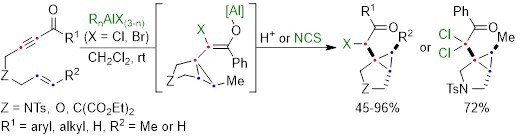
Oxidative Mizoroki–Heck reaction of unprotected cinnamylamines at ambient temperature under air
Olutayo N. Farinde, Vanaparthi Satheesh, Kendra K. Shrestha, Carmen R. Rhinehalt, Vinod G. Landge and Michael C. Young
Org. Chem. Front., 2023, 10, 3982-3988
Cinammylamines and 3,3-diarylallylamines represent an important class of molecules for drug discovery, and can serve as intermediates thanks to their olefin functionality which can lead to further elaboration. However, there are numerous challenges in the application of organometallic methods to synthesize highly-functionalized 3,3-diarylallylamines. Historically these reactions required protecting groups, with the protecting group both limiting substrate decomposition as well as improving regioselectivity by imparting a directing effect (Scheme 1a). Recent efforts have established feasibility for direct free amine-directed reactions that avoid the need for protection and deprotection, but which suffer from selectivity issues due to the presence of different Pd species which promote different reaction pathways (Scheme 1b). Notably, β-protio allylamines undergo both Heck reactions to give trans-functionalised products as well as C–H activation to give the cis-functionalised products as mixtures. Meanwhile, β-substituted allylamines preferentially undergo a γ,γ’-diarylation reaction rather than the simple γ-monoarylation due to the reactivity of the in situ-formed Pd nanoparticles responsible for the reaction.
To solve the stereoselectivity challenge, the group of Prof. Michael C. Young at the University of Toledo has unveiled a new protocol for selective Mizoroki–Heck arylation of unprotected cinnamyl and allylamines (Scheme 1c). Compared with previous work, a pivotal change was to exploit an oxidative Heck reaction using aryl boronic acid as the coupling partner. By using the aryl boronic acid, the conditions could be made sufficiently mild to essentially shut down the C–H activation pathway, giving >20:1 selectivity in most cases.
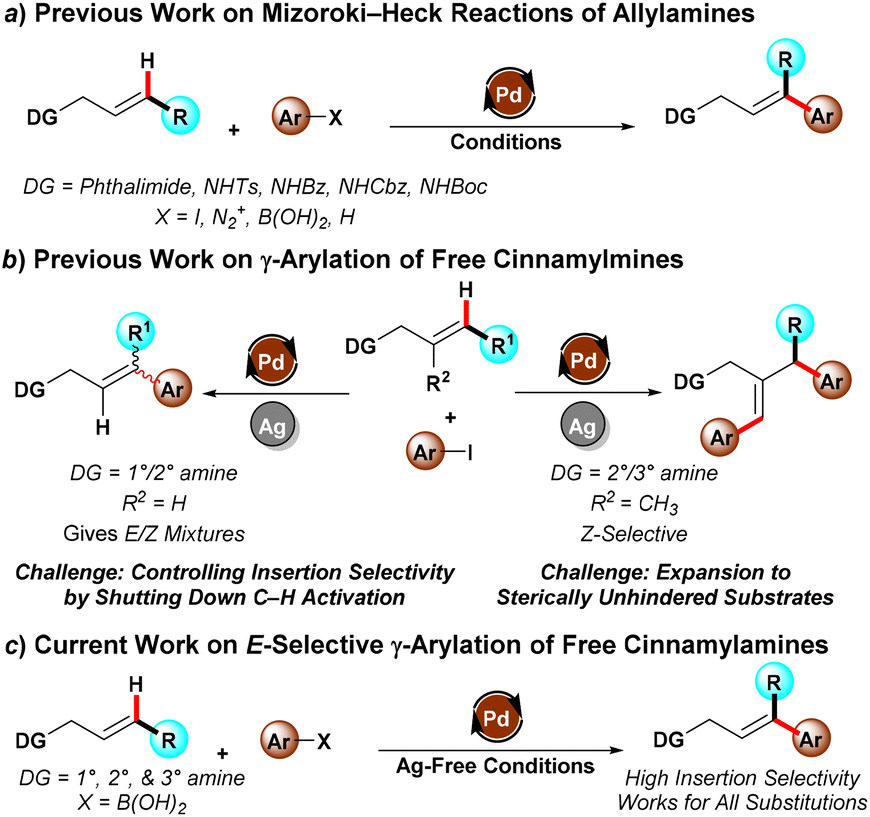
Scheme 1. Approaches for the Mizoroki–Heck reaction of allyl and cinnamylamines.
The researcher team demonstrated the broad applicability of their approach by successfully synthesizing a range of unsymmetrical 3,3-diarylallylamine derivatives with overall good structural diversity. The method presented by the research team not only improves the overall synthetic process but also offers a pathway to access a wider range of drug molecules in a more cost-effective manner. By comparing their reimagined Mizoroki–Heck reaction with existing methodologies utilizing protected amine substrates and aryl iodides, the researchers highlighted the numerous advantages of their approach. With improved atom and step economy, higher selectivity, and broader substrate scope, their method represents a significant advancement in the field of organic synthesis.
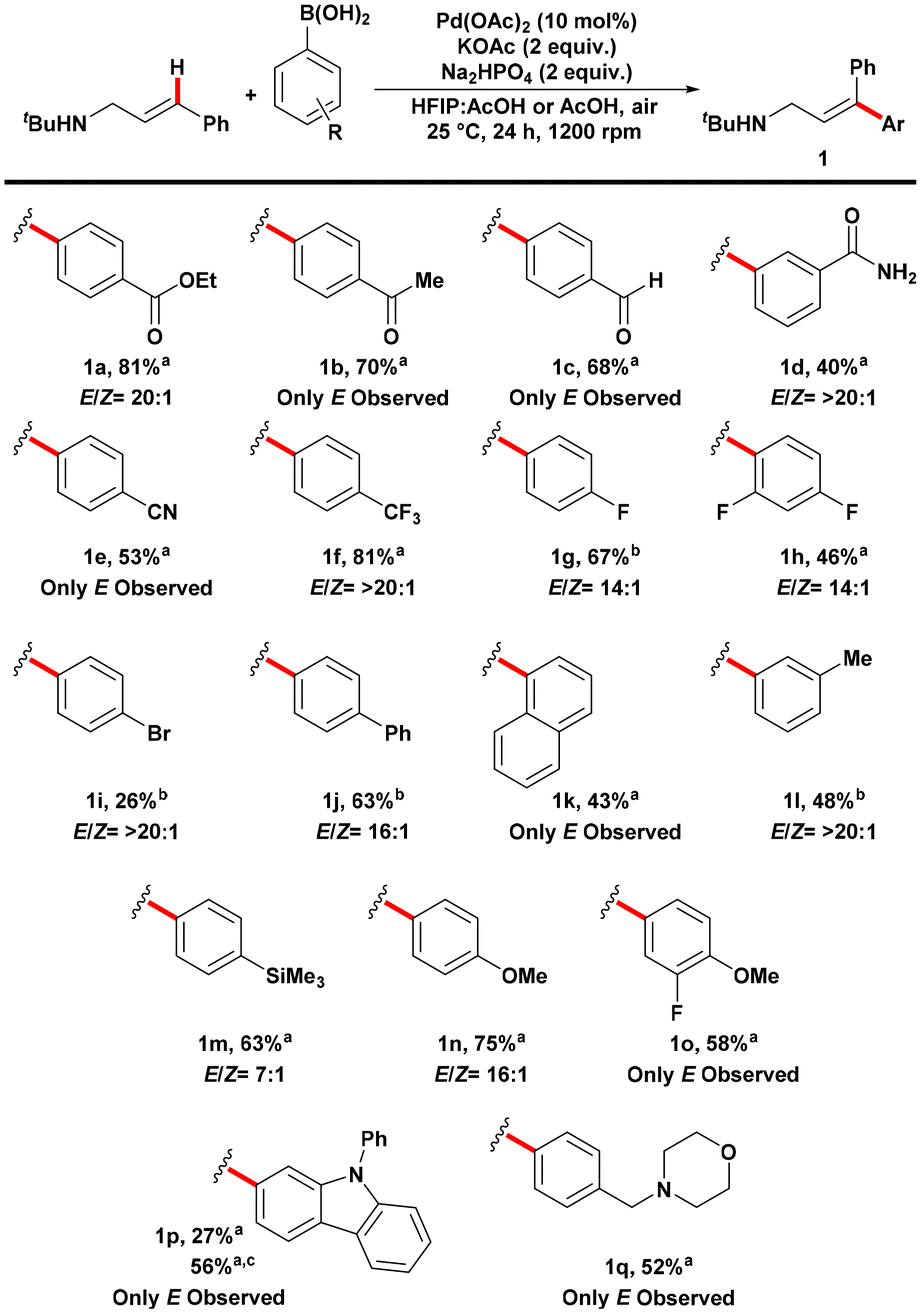
Scheme 2. Substrate scope of aryl boronic acid coupling partners in the E-selective Mizoroki–Heck reaction on cinnamylamines. Reactions were performed using amine (0.15 mmol) and aryl boronic acid (0.30 mmol) under air atmosphere. All reactions were performed in triplicate and the average isolated yields reported. E/Z ratios were determined from the crude 1H NMR using 1,1,2,2-tetrachloroethane as internal standard. a Solvent was 0.9 mL HFIP:0.3 mL AcOH. b Solvent was 4.0 mL AcOH. c Reaction was performed under 1 atm of O2.
The researchers examined electron-deficient carbonyl-containing compounds with various functional groups, such as esters, ketones, aldehydes, amides, and nitriles (Scheme 2). The reaction proceeded successfully for all these compounds, and the nitrile group remained intact without undergoing hydrolysis, unlike in previous harsher conditions using a different coupling partner. The reactions generally exhibited high E-selectivity, with the Z-product undetectable in several cases. The researchers also explored electron-poor substrates containing fluorides and bromides, although the bromide compound had a lower yield. The recovery of the starting amine was reasonably high, and analysis revealed that the low yield of the bromide compound was due to a complex mixture of products resulting from Suzuki-Miyaura cross-coupling. Conjugated systems and electron-rich groups on the arene were also compatible with the reaction. Additionally, a coupling partner with both electron-withdrawing and donating groups yielded the desired product without the formation of the Z-isomer.
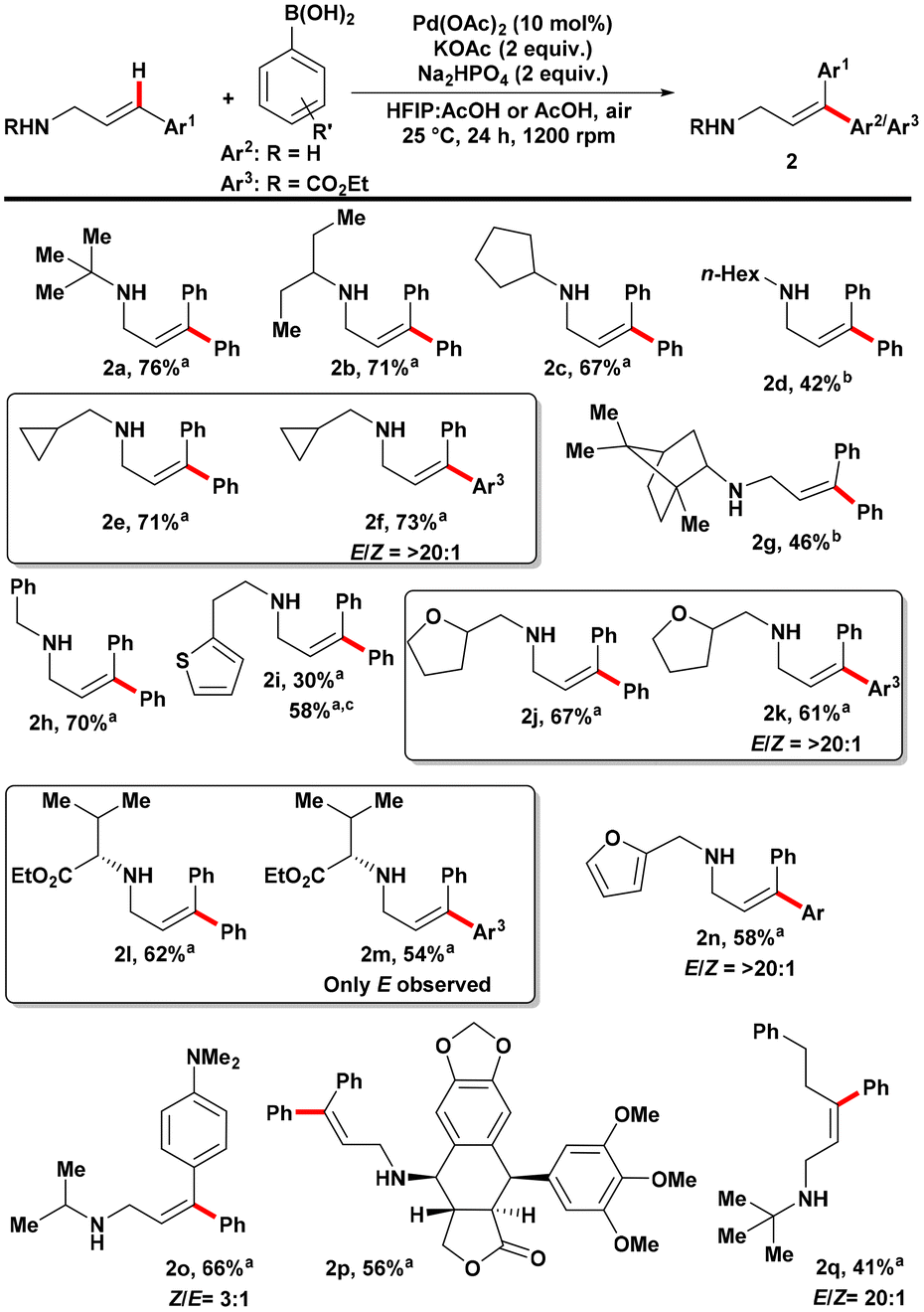
Scheme 3. Substrate scope of cinnamylamine coupling partners in the E-selective Mizoroki–Heck reaction on cinnamylamines. Reactions were performed using amine (0.15 mmol) and phenyl boronic acid (0.30 mmol) under air atmosphere. All reactions were performed in triplicate and the average isolated yields reported. E/Z ratios were determined from the crude 1H NMR using 1,1,2,2-tetrachloroethane as internal standard. a Solvent was 0.9 mL HFIP:0.3 mL AcOH. b Solvent was 4.0 mL AcOH. c Reaction was performed under 1 atm of O2.
In the investigation of amine substrate scope, various secondary amines were found to be viable substrates (Scheme 3). This included secondary amines with α-tertiary, α-secondary, and α-primary aliphatic substituents. Although cyclopropylamine showed low reactivity, introducing a methylene spacer enabled the generation of a reactive substrate. When targeting the addition of phenyl groups to the substrate, a spot check revealed a strong preference for the E-product over the Z-product (>20:1 ratio) when using 4-ethoxycarbonyl phenylboronic acid as the coupling partner. Sterically congested bornylamine derivatives, aliphatic amines with aromatic or heteroaromatic groups, and saturated heterocycles such as tetrahydrofurfuryl could also be successfully converted to the desired products. Similar E/Z-selectivity was observed when using 4-ethoxycarbonyl phenylboronic acid as the coupling partner for these substrates. Amino acid-derived substrates yielded good yields of the product, with complete E-selectivity observed based on the crude reaction mixture’s 1H NMR. The stereochemistry of the amino ester was confirmed to have >99% enantiomeric excess (ee) based on Mosher amide analysis. Electron-rich furfurylamine-derived substrates were viable, whereas pyridines did not show reactivity. N-tert-butyl-(4,4-dimethylamino)cinnamylamine, which decomposed under previous harsh conditions, could be converted to the desired product under the milder conditions, albeit with decreased Z/E-selectivity. Furthermore, an amine derived from podophyllotoxin, containing several oxygen-based functional groups, successfully participated in the reaction. Control of β-hydride elimination was achieved when a 3-(phenethyl)-substituted allylamine was used, resulting in the formation of the β,γ-unsaturated allylamine product. Expanding the scope to less and more-substituted substrates, primary cinnamylamine was functionalized with the desired product in 68% yield. The need for CO2 to prevent amine decomposition during the reaction, as in previous work, was not necessary under the milder conditions. Acyclic and cyclic amine substrates could be functionalized with good yields, demonstrating the versatility of this modified approach to amine functionalization. Trans-selectivity was observed when unsymmetrical 3,3-diarylallylamines were used as substrates.
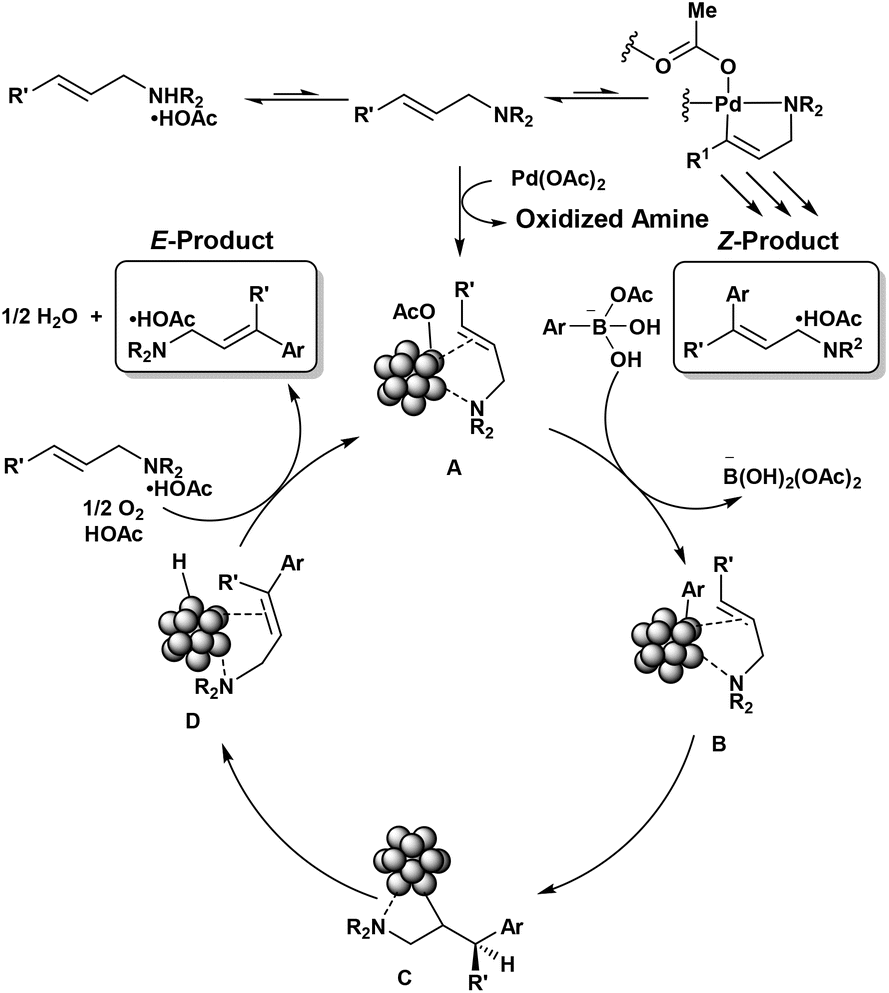
Figure 1. Proposed catalytic cycle.
Young and his coworkers propose a preliminary mechanistic understanding of the Mizoroki-Heck reaction using aryl boronic acids as coupling partners (Figure 1). They suggest that the active catalyst is formed through the incomplete reduction of Pd(OAc)2 to Pd nanoparticles (NPs) ligated by allylamine. Transmetallation with activated aryl boronic acid occurs through ligand exchange, followed by C–Pd bond insertion across the alkene. This forms an organic metallic intermediate, which undergoes β-hydride elimination to yield the desired product coordinated to the nanoparticle. The catalyst is regenerated through oxidation of the metal-hydride in the presence of molecular oxygen and acetic acid, while the functionalized amine is exchanged for unfunctionalized substrate. Further investigations are needed to confirm and refine this proposed mechanism.
The developed method not only demonstrates compatibility with 1°, 2°, and 3° amines but also showcases its ability to facilitate chain walking reactions. Moreover, it provides practical and scalable solutions to address longstanding challenges in drug synthesis. The implications of this research extend far beyond the laboratory, as it opens up new possibilities for pharmaceutical development. By harnessing the power of the Mizoroki–Heck reaction with aryl boronic acids, researchers can now expedite the synthesis of 3,3-diarylallylamines, ultimately accelerating the discovery and development of novel drugs targeting G protein-coupled receptors. This breakthrough has the potential to revolutionize the pharmaceutical industry, improving drug accessibility and affordability for patients worldwide. Its impact on drug synthesis and the potential for transforming the pharmaceutical landscape cannot be overstated. This innovative approach sets the stage for further advancements in the field and offers hope for the discovery of new and more effective medications.
 Prof. Michael Christopher Young |
Michael Young was born in King, N.C. (approximately where Mayberry of Andy Griffith fame would be if it were real). He always knew he wanted to be a scientist, and while studying at Western Carolina University decided that chemistry was a better career choice than trying to genetically engineer Pokémon. He began his independent career after a PhD (2014) at UC – Riverside with Prof. Richard Hooley and a postdoc (2014 – 2016) at UT – Austin with Prof. Guangbin Dong. His group is interested in developing more sustainable routes to biologically relevant compounds, with a particular interest in G-protein coupled receptor-agonists and antagonists. |
 Olutayo Nathanael Farinde |
Olutayo Nathanael Farinde is a PhD student currently working in the Young lab at The University of Toledo. He holds a BS degree in Analytical and Environmental Chemistry from the University of Ibadan, Nigeria, which he completed in 2019. At The University of Toledo, Nate’s research focuses on the area of C–H functionalization and alkene functionalization of amines. He is interested in developing novel synthetic methods which can be used to synthesize biologically-active molecules. When he’s not at the bench, Nate enjoys learning new computational methods.
|
 Dr. Satheesh Vanaparthi |
Satheesh Vanaparthi was born and raised in Telangana, India. He obtained his BSc and MSc from Osmania University and later received his PhD from Indian Institute of Technology – Guwahati, India (Prof. T. Punniyamurthy, 2019). After a postdoc at the Technion-Israel Institute of Technology, Israel (Prof. Graham de Ruiter), Satheesh moved to The University of Toledo, USA to work with the Young lab where he is currently working on rhodium and palladium-catalysed transformations. |
 Kendra Kumar Shrestha |
Kendra hails from Nepal. He obtained his BSc and MSc from Tribhuvan University. In 2019, he moved to The University of Toledo to pursue his Ph.D. with Prof. Michael C. Young where he is working on palladium-catalysed selective functionalisations of aminees as wel as cycloaddition reactions exploiting organo-supramolecular catalysis. |
 Carmen Rose Rhinehalt |
Carmen is a chemistry major with a passion for how and why the world works. She is currently working on the deuteration of benzoic acid derivatives and would like to transfer everything she has learned in the lab and chemistry to the development of cosmetic products. In her free time, Carmen likes to try new recipes and bake all kinds of sweets! |
 Dr. Vinod Gokulkrishna Landge |
Vinod obtained his PhD from the National Chemical Laboratory in Pune, India, in 2019, where worked with Prof. Ekambaram Balaraman. He then joined the Young lab where he worked extensively on Pd and organocatalysis. He current works as a Process Development Scientists at Piramal Pharma Solutions. |








")