Thermodynamic- and kinetic-controlled deprotonation of symmetric and asymmetric ketones is a well-known process that leads to the formation of enolates, one of the most important intermediate in organic chemistry. Regio-selective deprotonation assumes extreme importance, being critical to the success of organic reactions, when unsymmetrical carbonyl compounds such as ketones, having α-hydrogens in both sides, are taken into account. To the best of our knowledge, no studies have specifically reported on β-functionalized ketones in which regioselective formation of enolates could be less obvious and intuitive with respect to that observed in the most familiar α-functionalized analogues.
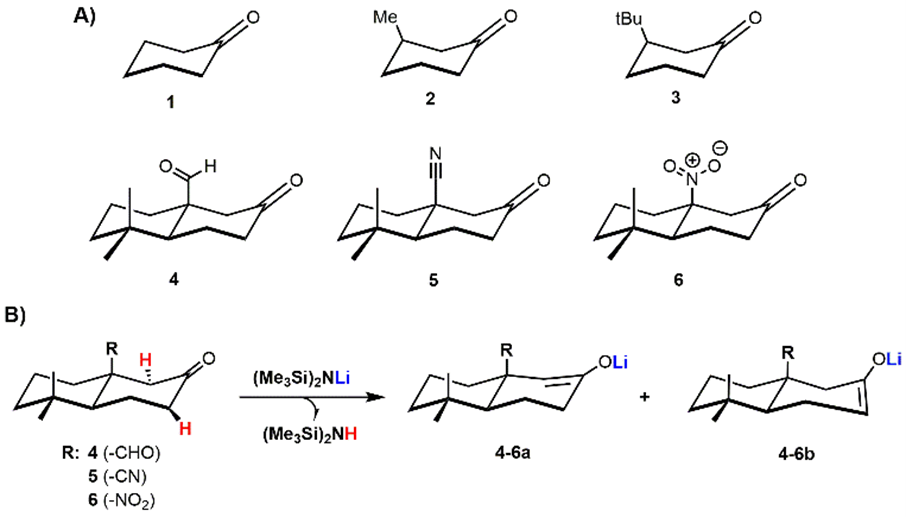
Figure 1. Chemical structure of ketones 1–6 involved in this study (A). Potential thermodynamic (4–6a) and kinetic enolates (4–6b) from the deprotonation of 4–6 using LHMDS (B).
Here we report a theoretical study at the DFT level of theory on a group of β-functionalized cyclic ketones 1–6 (Figure 1) to shed light on kinetic parameters (e.g., electronic effect and steric hindrance) and any other parameters (e.g., non-covalent interactions) related to the formation of kinetic and thermodynamic enolates through the use of hindered and strong bases, such as LHMDS and KHMDS. These bases have been taken into account as models to investigate the effect of counter cations (Li+ or K+) on the enolate formation process.
Surprisingly, “thermodynamic enolate” 4a was obtained as single product under kinetic conditions (THF, −78 °C) while ketone 5, gives “kinetic product” 5b as main enolate under same conditions with a regioselectivity of 70 : 30 over the “thermodynamic product” 5a. The unusual behavior reported in the literature for ketones 4 and 5 in the presence of LHMDS captured our attention and fed our curiosity.
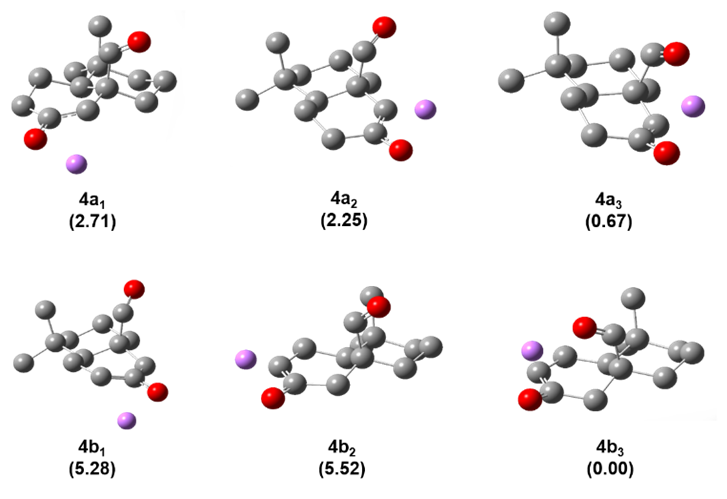
Figure 2. Conformers and relative stability from Li-enolates from ketone 4. Relative stability for each Li-conformers is reported in kcal/mol in parentheses. Hydrogens have been hidden for better clarity.
A conformational study on the Li+@4 complex highlights the coordination ability of the functional group in β-positon (e.g. aldehyde group) and unveils its effect on the resulting enolate distribution (see Figure 2). The lack of functional groups with coordination ability in 1–3 does not allow the double coordination of Li+ cation and consequently, no alteration of the enolate distribution is observed when (Me3Si)2– was exchanged with LHMDS. The observed effect must be attributed to the coordination ability of the functional group (e.g. aldehyde in 4, cyano in 5 and nitro group in 6) and their directionality. Aldehyde and nitro group in 4 and 6, respectively, showed a superior directionality compared to cyano group in 5, leading to a higher stabilization of the relative TS for the presence of Li+ or K+ cations and consequently, higher regioselectivity towards “thermodynamic enolate” as experimentally reported (Table 1).
Table 1 Activation energy (Ea), ∆EEa and ∆∆EEa for ketones 4–6 in the presence of Li+ and K+.
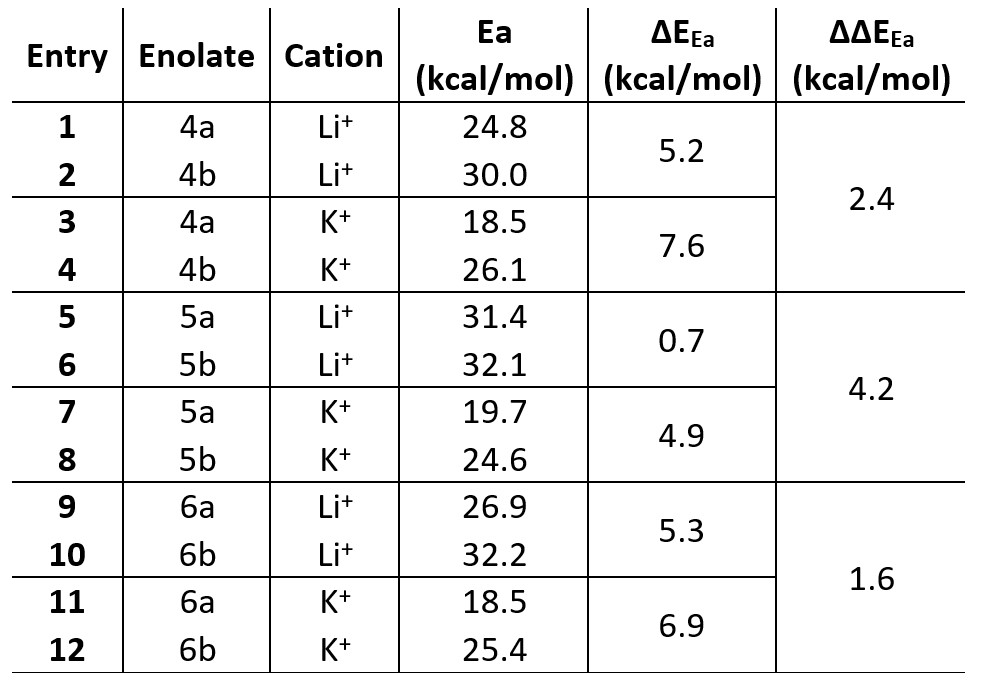
On the other hand, the linear orientation of the cyano group (–C≡N) in 5 does not allow a proper coordination with the Li+ cation, showing a ∆EEa of 0.7 kcal/mol and leading us to suppose the presence of both enolates (5a and 5b) in the resulting solution (see Table 1 – Entries 5 and 6) as reported in literature. However, higher regioselectivity towards the “thermodynamic enolate” 5a is detected in presence of KHMDS where a ∆EEa of 4.9 kcal/mol is now observed (see Table 1 – Entries 7 and 8). This is rationalized by the better coordination between K+ cation and cyano group derived from the bigger size of the K+ cation with respect to the Li+ cation (Figure 3), highlighting how preliminary base selection (e.g., LHMDS vs. KHMDS) could strongly affect the resulting enolate distribution.
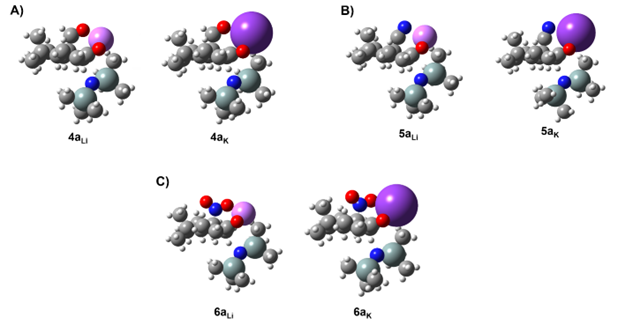
Figure 3. Comparison between thermodynamic enolates 4a (A), 5a (B) and 6a (C) coordinated to Li+ (left) and K+ (right).
We believe this work: (i) sheds light on the kinetic parameters that affect the regioselectivity in β-functionalized cyclic ketones, (ii) gives reasonable explanations for the unusual regioselectivity reported in the literature for some β-functionalized cyclic ketones such as 4 and 5, and (iii) could be a source of inspiration for future regioselective synthetic approaches and promotes the use of non-covalent interactions in the field of stereochemistry.
Corresponding author:









")