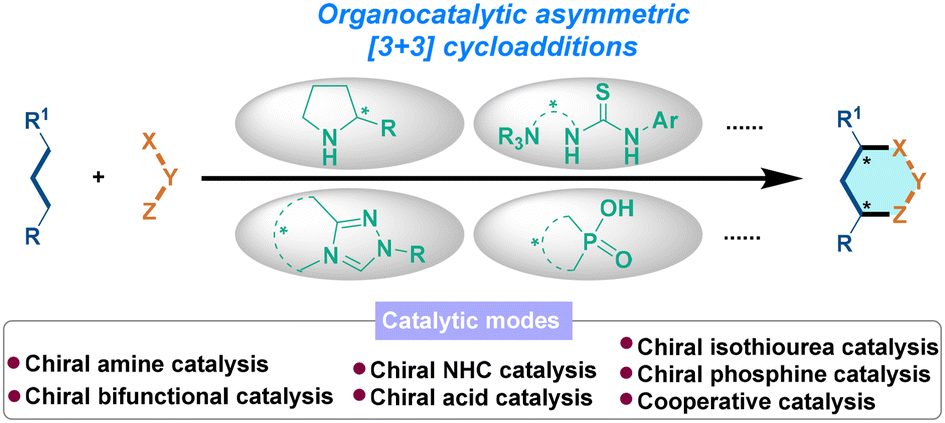
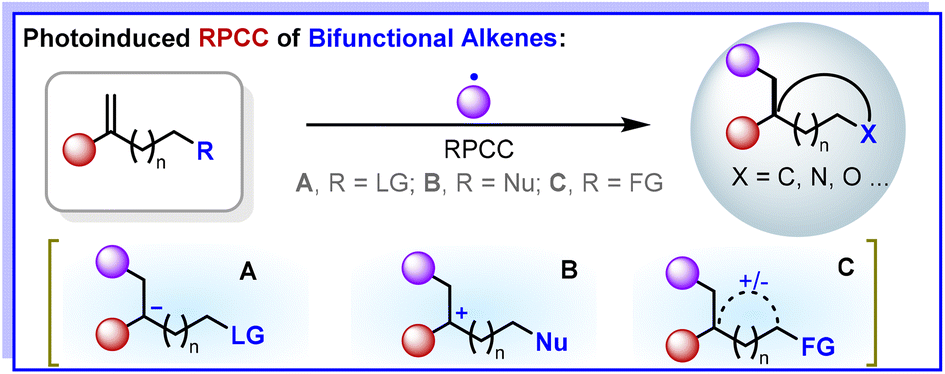
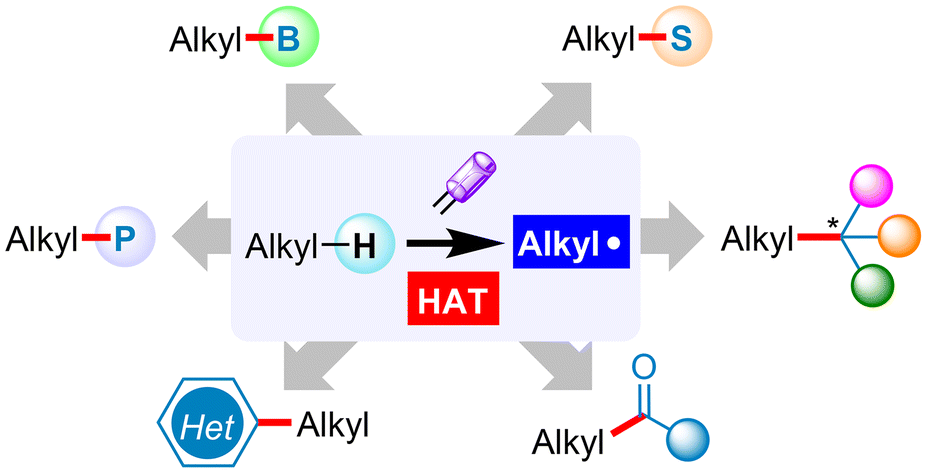
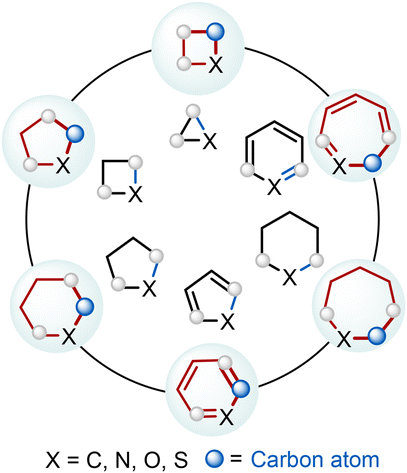
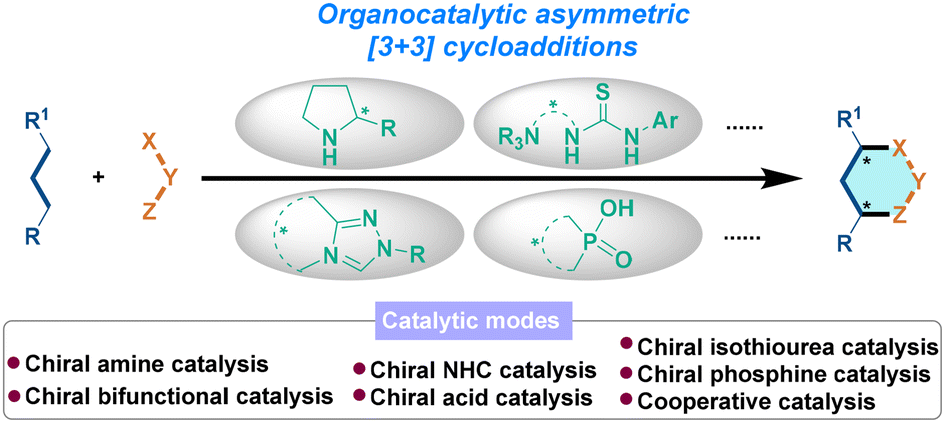
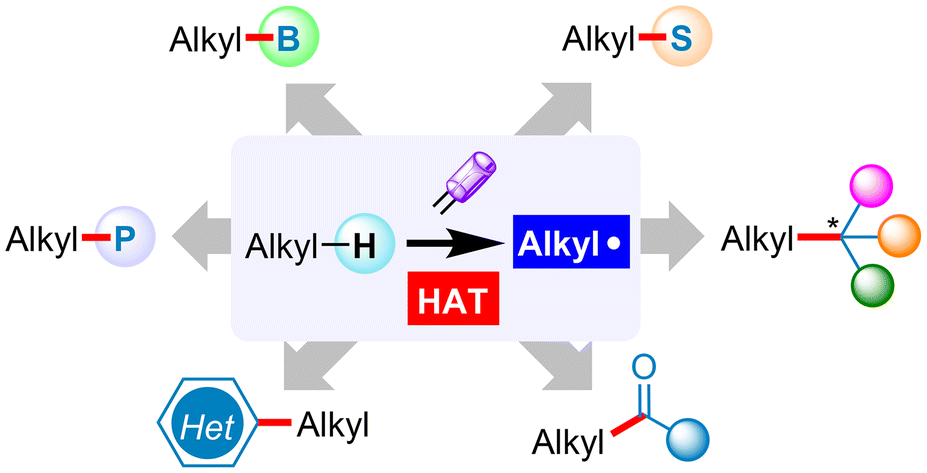
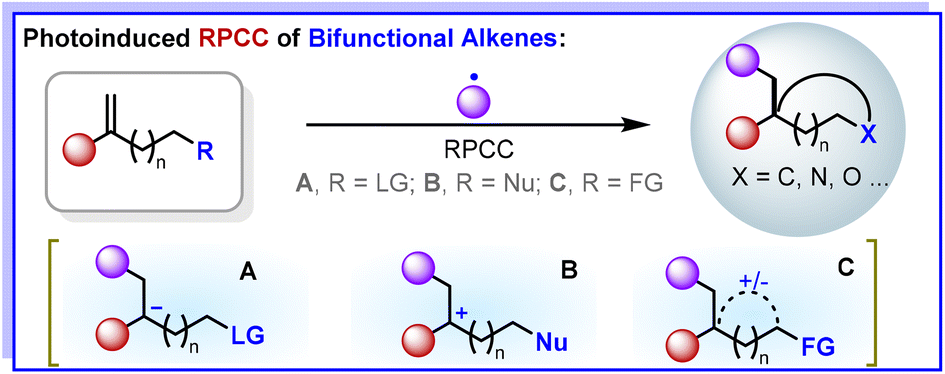
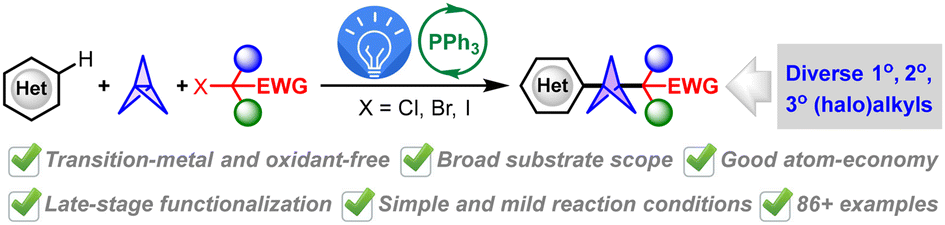
We are delighted to highlight the following highly cited research articles published in Organic Chemistry Frontiers since 2023. We hope you enjoy reading them.
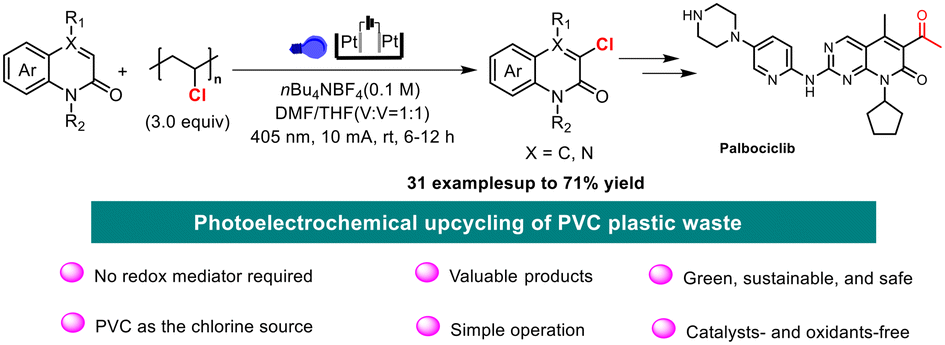 |
Photoelectrochemical upcycling of PVC plastic waste for the synthesis of chlorinated quinolinone derivatives
Kai Zheng, Jingwen He, Lixi Zhang, Zhaoyang Wang, Pengfei Zhang, Jianyu Cao and Chao Shen Org. Chem. Front., 2025,12, 3191-3197 |
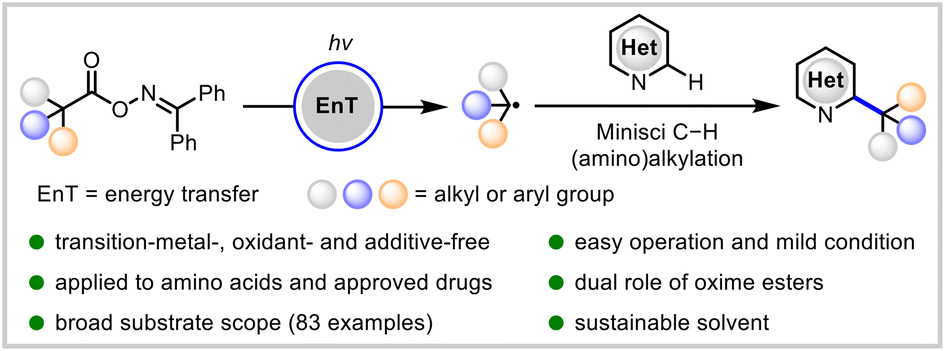 |
Energy-transfer photocatalysis for Minisci C–H (amino)alkylation of heteroarenes using oxime esters as dual-role reagents
Jun Xu, Yuru Zhang, Ruiyuan Xu, Yuxin Wang, Jiabin Shen and Wanmei Li Org. Chem. Front., 2024,11, 5122-5129 |
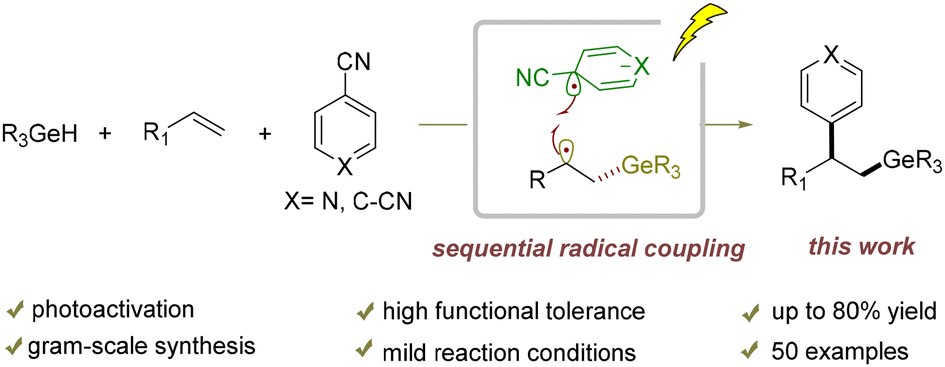 |
Arylgermylation of alkenes by a cooperative photoactivation and hydrogen atom transfer strategy
Jia Cao, Yan Liu, Zhixiang Wang and Le Liu Org. Chem. Front., 2024,11, 7098-7106 |
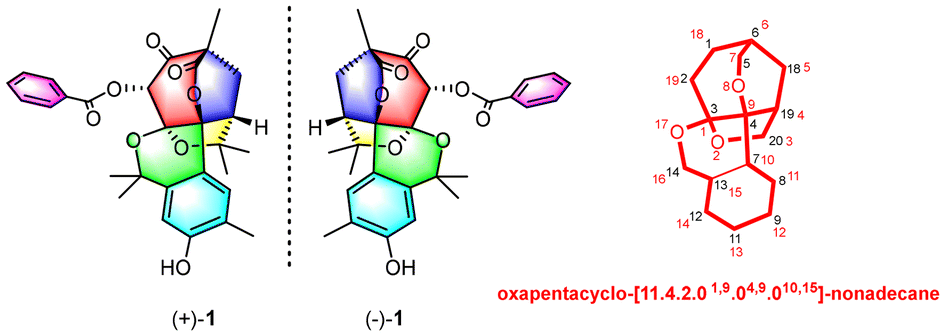 |
(±)-Hypandrone A, a pair of polycyclic polyprenylated acylphloroglucinol enantiomers with a caged 7/6/5/6/6 pentacyclic skeleton from Hypericum androsaemum
Jiangchun Wei, Pingping Fan, Yahui Huang, Hanxiao Zeng, Rui Jiang, Zhengzhi Wu, Yonghui Zhang and Zhengxi Hu Org. Chem. Front., 2024,11, 3459-3464 |
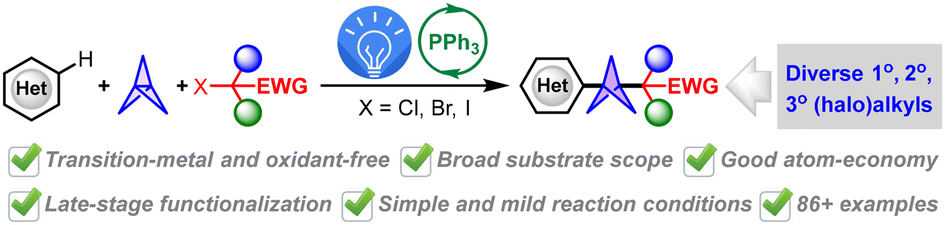 |
Transition-metal-, oxidant- and additive-free multi-component synthesis of alkyl heteroaryl BCPs enabled by visible-light-induced phosphine-catalyzed halogen-atom transfer
Jun Xu, Yu Hong, Ruiyuan Xu, Yuxin Wang, Yirui Guo, Jiabin Shen, Yuxuan Zhao and Wanmei Li Org. Chem. Front., 2024,11, 6166-6176 |
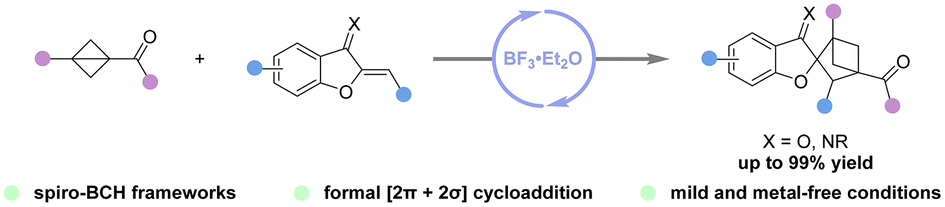 |
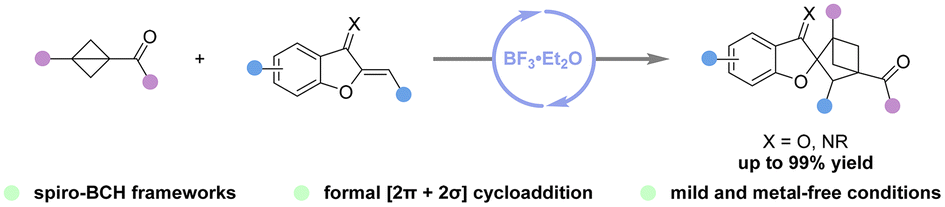
Access to spiro-bicyclo[2.1.1]hexanes via BF3·Et2O-catalyzed formal [2π + 2σ] cycloaddition of bicyclo[1.1.0]butanes with benzofuran-derived oxa(aza)dienes
Jia-Yi Su, Jian Zhang, Zhi-Yun Xin, Hao Li, Hanliang Zheng and Wei-Ping Deng Org. Chem. Front., 2024,11, 4539-4545 |
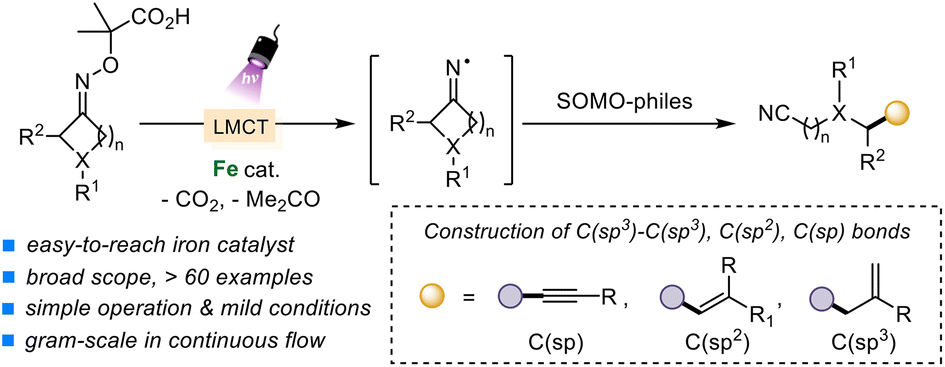 |
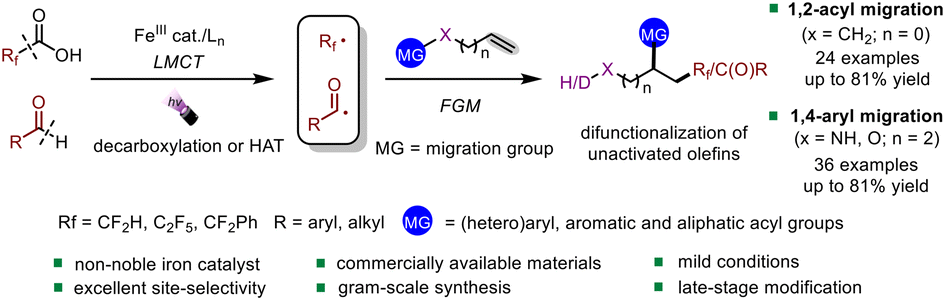
Iron-catalyzed fragmentation-alkynylation, -alkenylation and -alkylation cascade enabled by photoinduced ligand-to-metal charge transfer
Yining Zhu, Han Gao, Jia-Lin Tu, Chao Yang, Lin Guo,Yating Zhao and Wujiong Xia Org. Chem. Front., 2024,11, 1729-1735 |
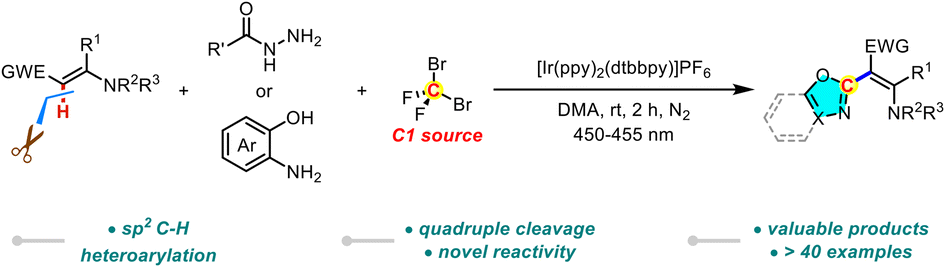 |
Photoinduced C–H heteroarylation of enamines via quadruple cleavage of CF2Br2
Wanqing Zuo, Lingling Zuo, Xiao Geng, Zhifang Li and Lei Wang Org. Chem. Front., 2023,10, 6112-6116 |
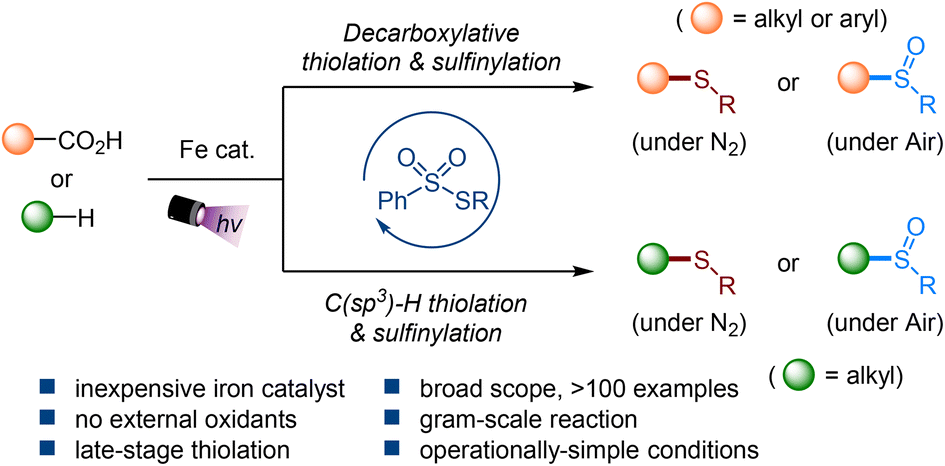 |
An iron-catalyzed C–S bond-forming reaction of carboxylic acids and hydrocarbons via photo-induced ligand to metal charge transfer
Ao-Men Hu, Jia-Lin Tu, Mengqi Luo, Chao Yang, Lin Guo and Wujiong Xia Org. Chem. Front., 2023,10, 4764-4773 |
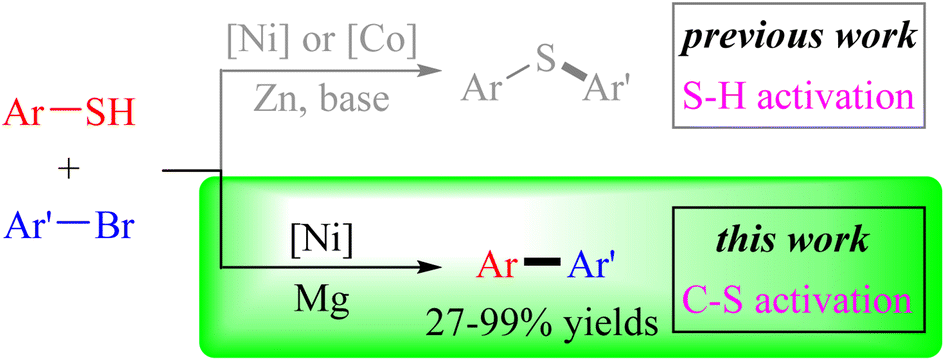 |
Nickel-catalyzed cross-electrophile coupling of aryl thiols with aryl bromides via C–S bond activation
Hao Xu, Cai-Yu He, Bo-Jie Huo, Jia-Wen Jing, Chengping Miao, Weidong Rao, Xue-Qiang Chu, Xiaocong Zhou and Zhi-Liang Shen Org. Chem. Front., 2023,10, 5171-5179 |








")