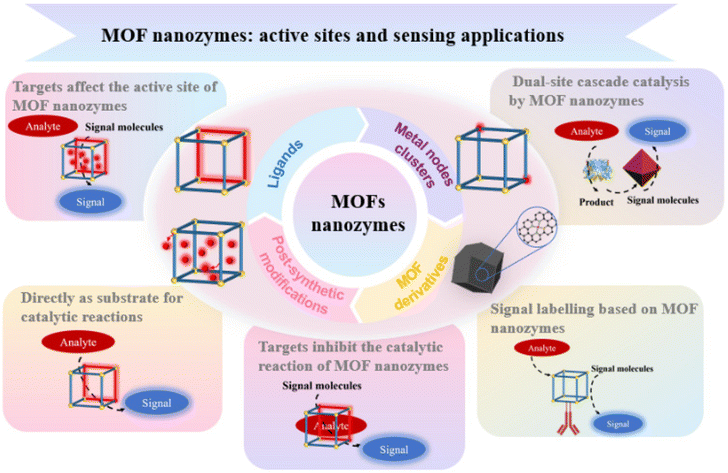
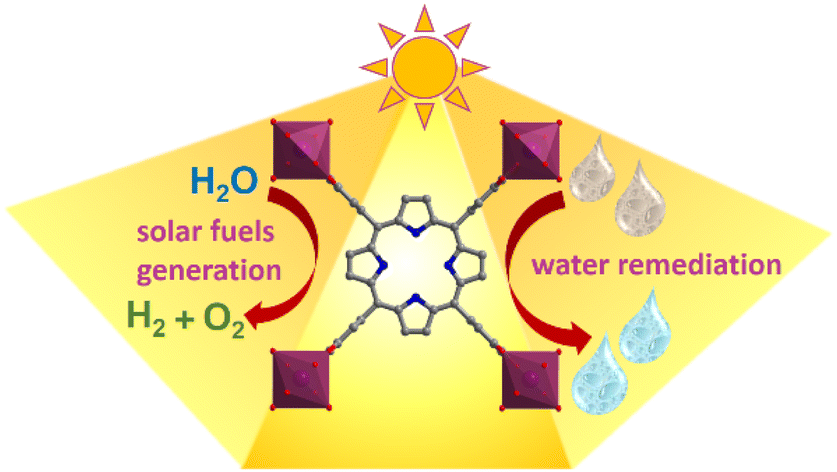
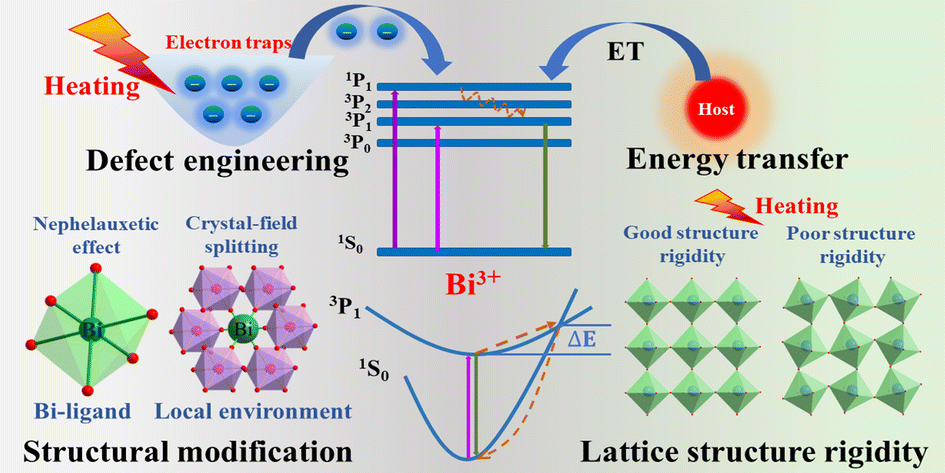
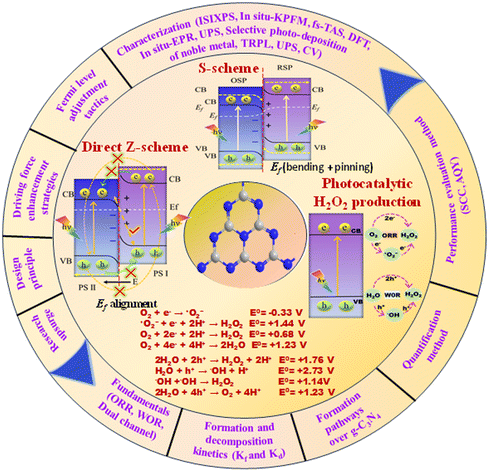
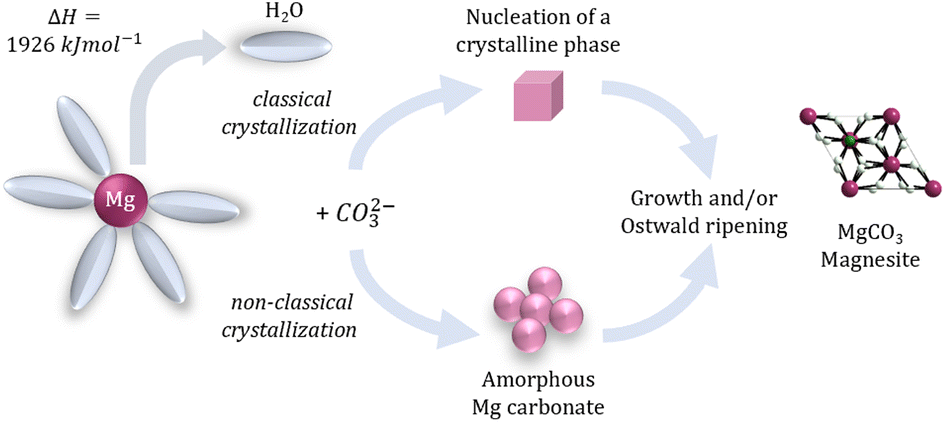
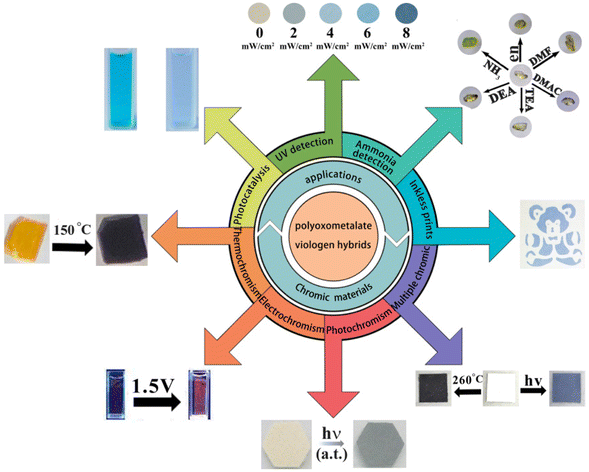
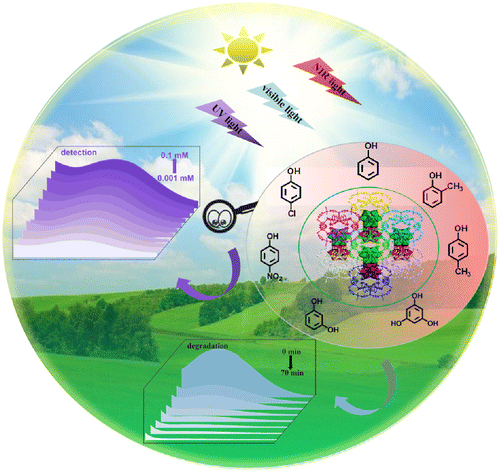
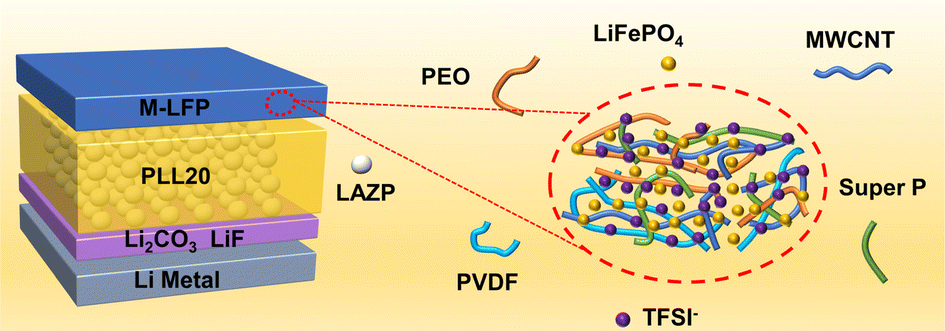
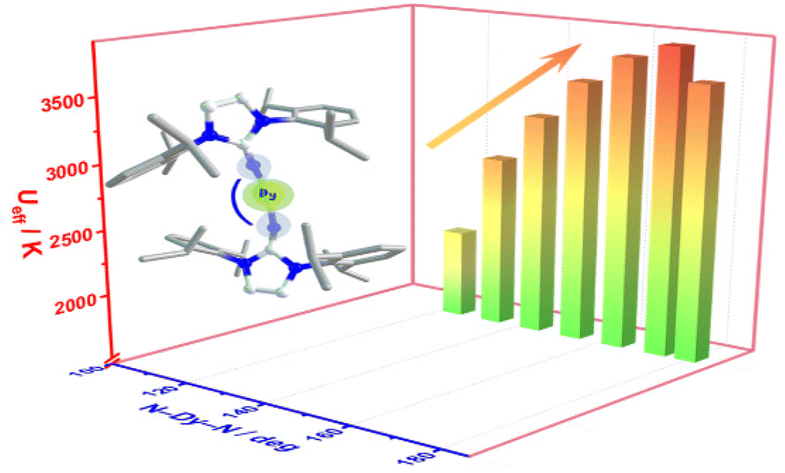
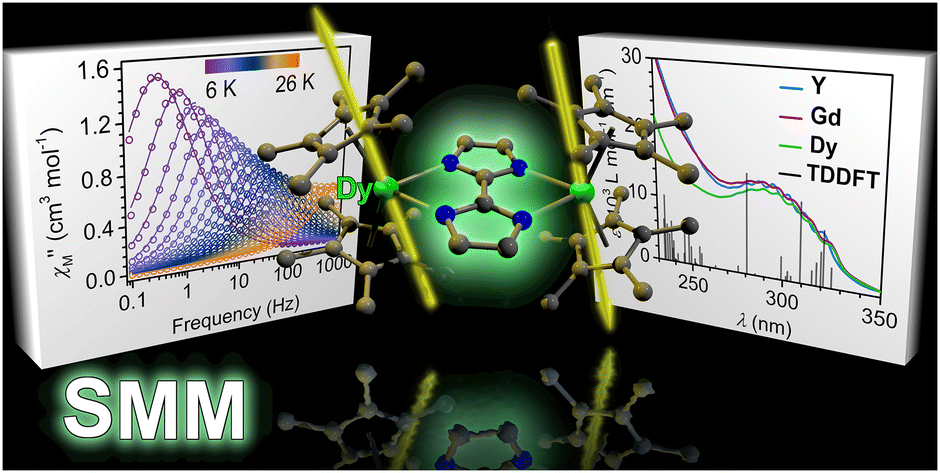
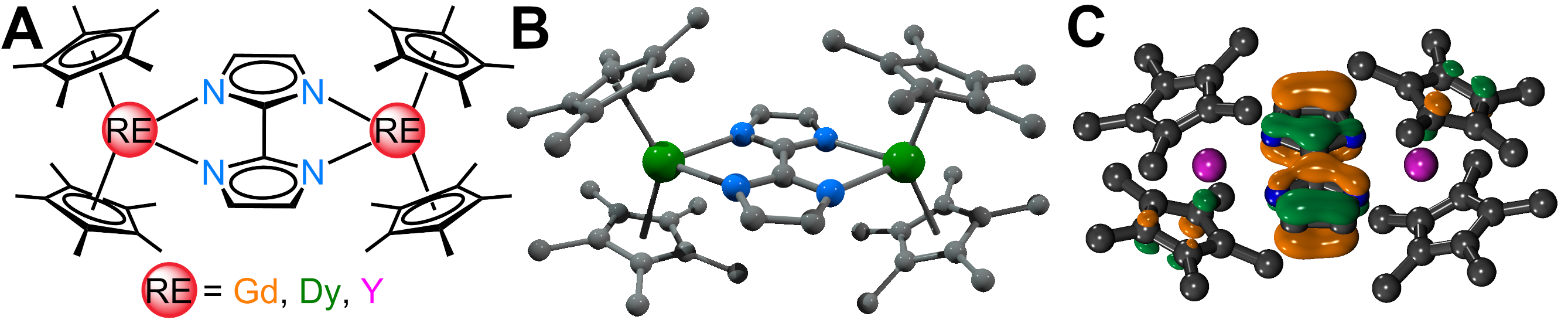
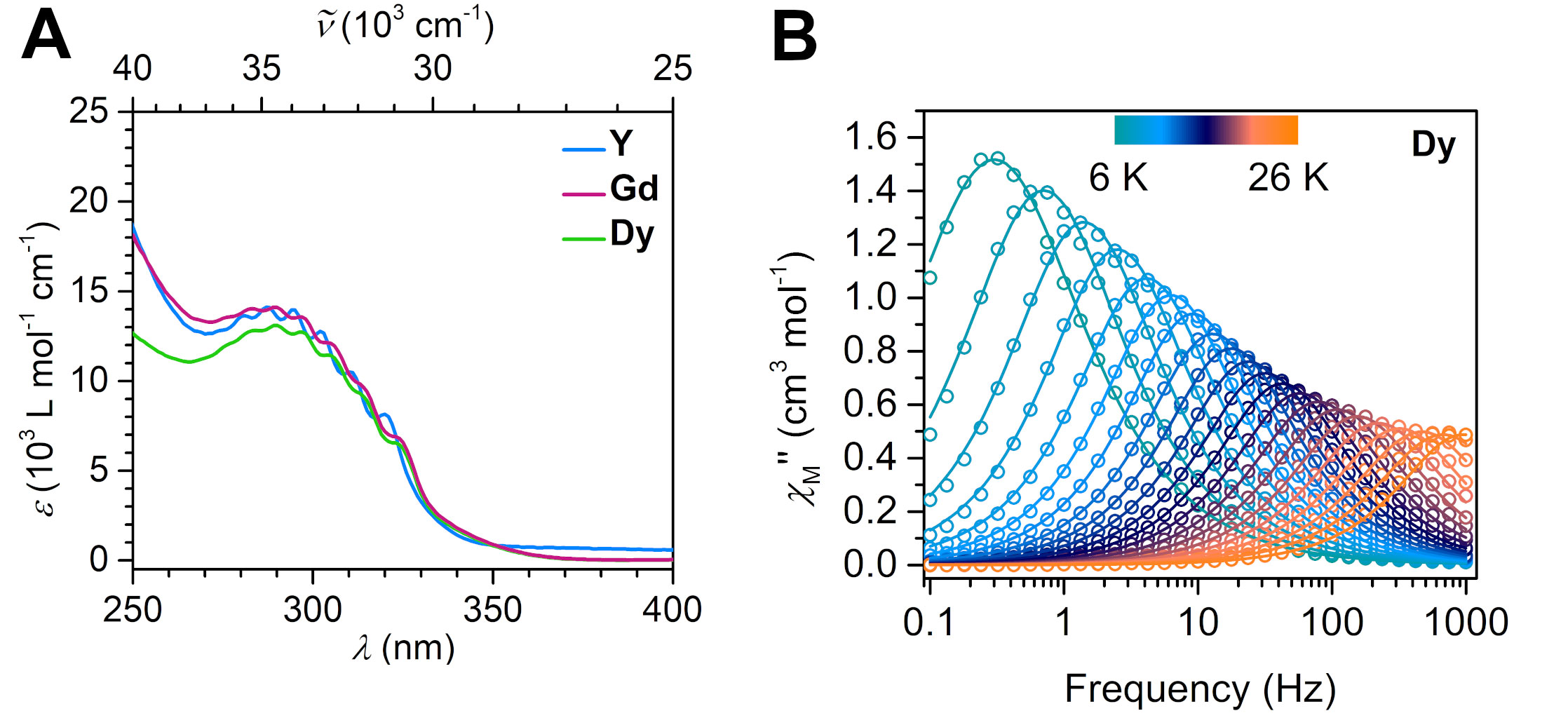
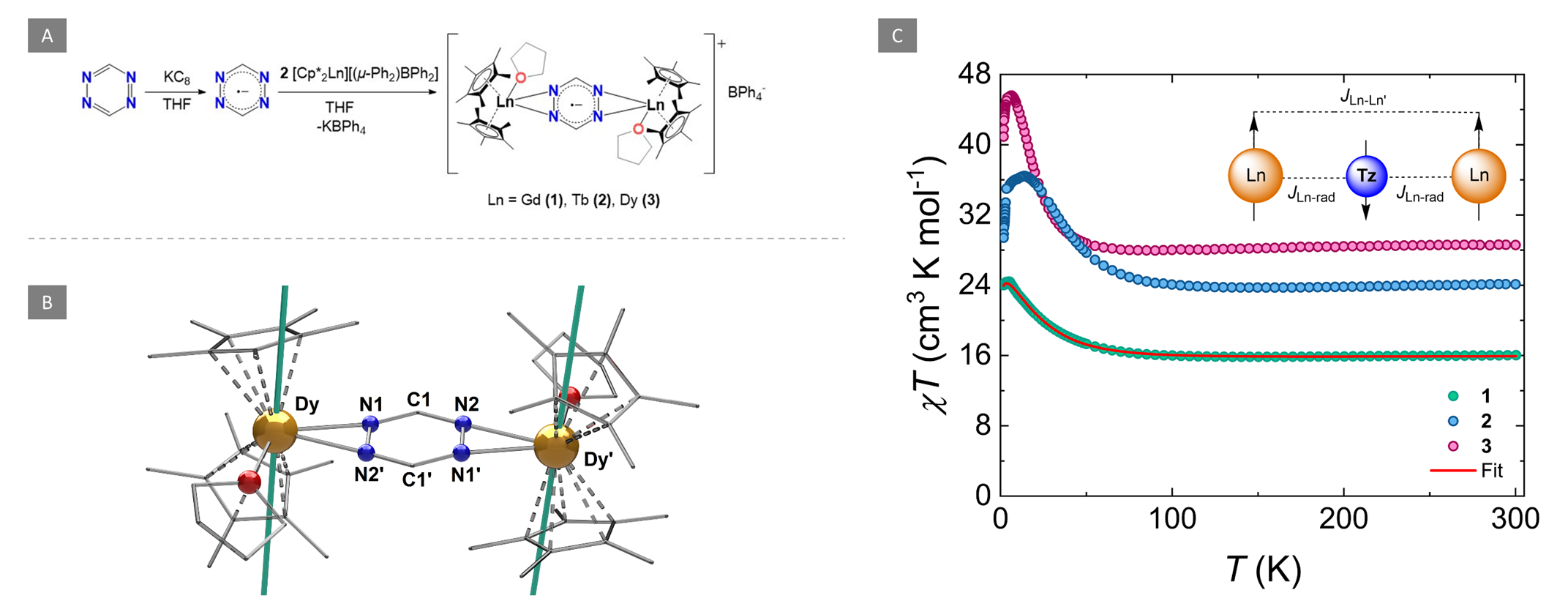
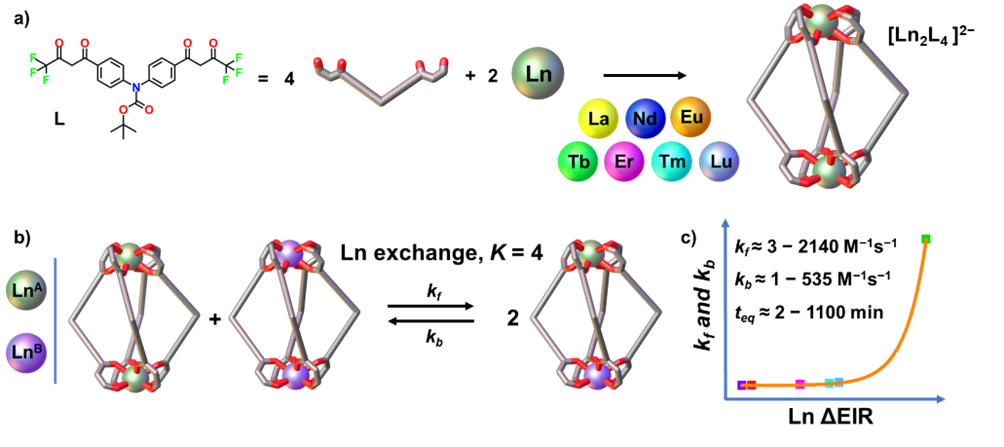
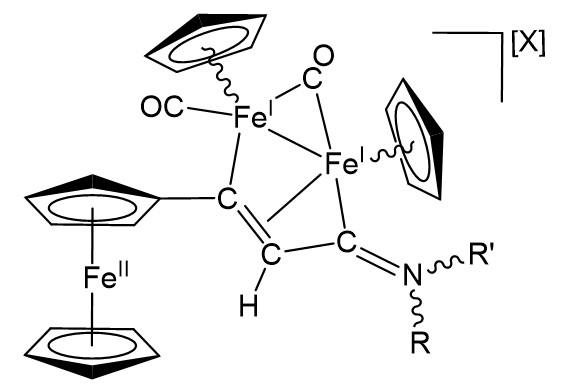
We are delighted to highlight the following highly cited research articles published in Inorganic Chemistry Frontiers since 2023. We hope you enjoy reading them.
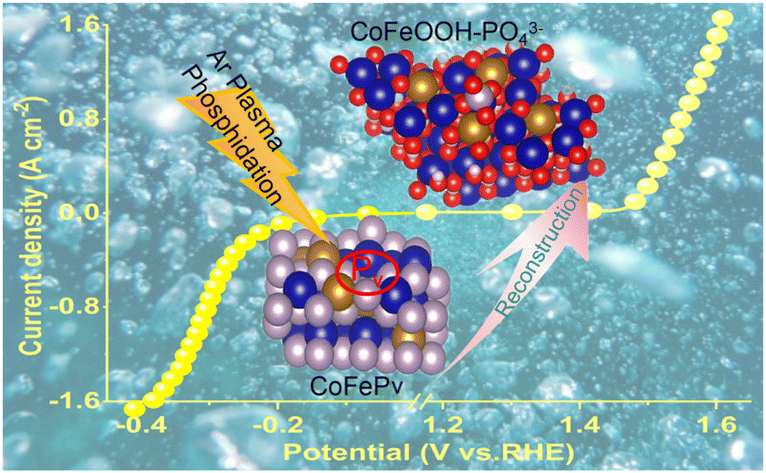 |
P vacancy-induced electron redistribution and phase reconstruction of CoFeP for overall water splitting at industrial-level current density
Inorg. Chem. Front., 2025,12, 2678-2690 |
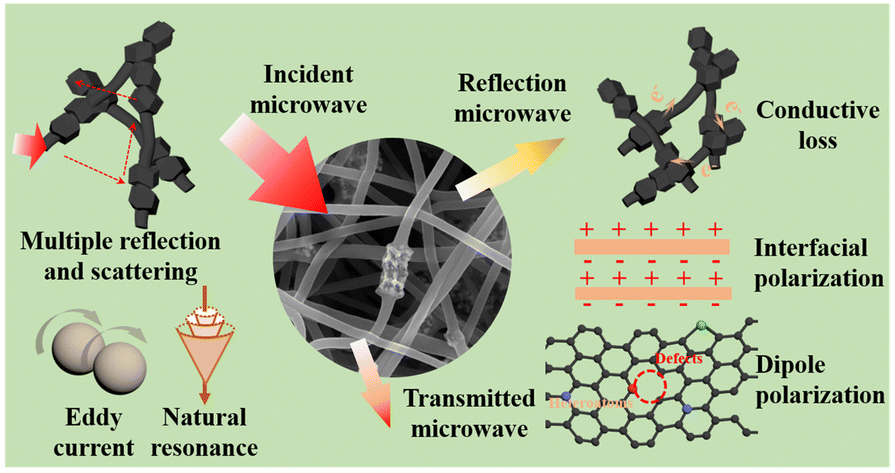 |
Enhanced interfacial polarization loss induced by hollow engineering of hollow alloyed CoFe-ZIF nanocages/carbon nanofibers for efficient microwave absorption
Dan Wu, Congmin Fan, Wusi Luo, Yingzhi Jin, Qinchuan He and Yiqun Wang |
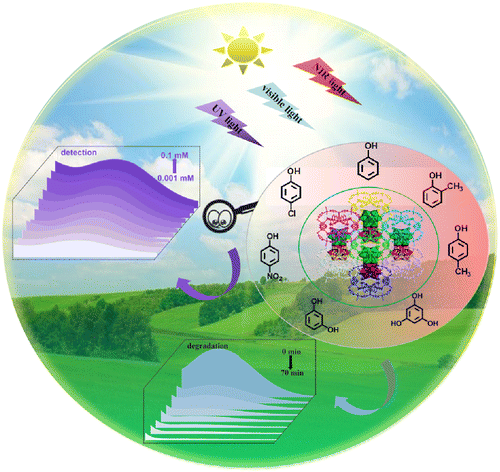 |
Achieving ultra-trace analysis and multi-light driven photodegradation toward phenolic derivatives via a bifunctional catalyst derived from a Cu(i)-complex-modified polyoxometalate
Shuang Li, Bingqian Wang, Guocheng Liu, Xiaohui Li, Chang Sun, Zhong Zhang and Xiuli Wang |
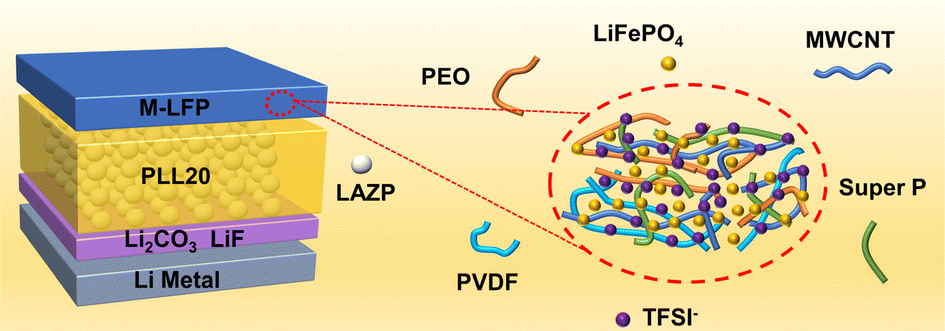 |
PEO/Li1.25Al0.25Zr1.75(PO4)3 composite solid electrolytes for high-rate and ultra-stable all-solid-state lithium metal batteries with impregnated cathode modification
Yongquan Zhang, Hongchang Gao, Jingshun Wang, Qingguo Chi, Tiandong Zhang, Changhai Zhang, Yu Feng, Yue Zhang, Dianxue Cao and Kai Zhu |
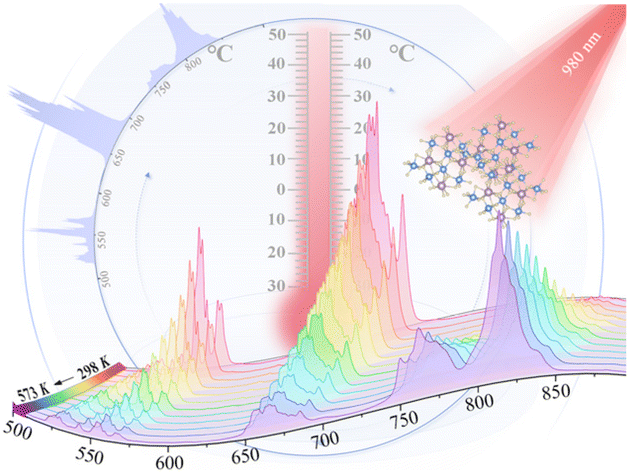 |
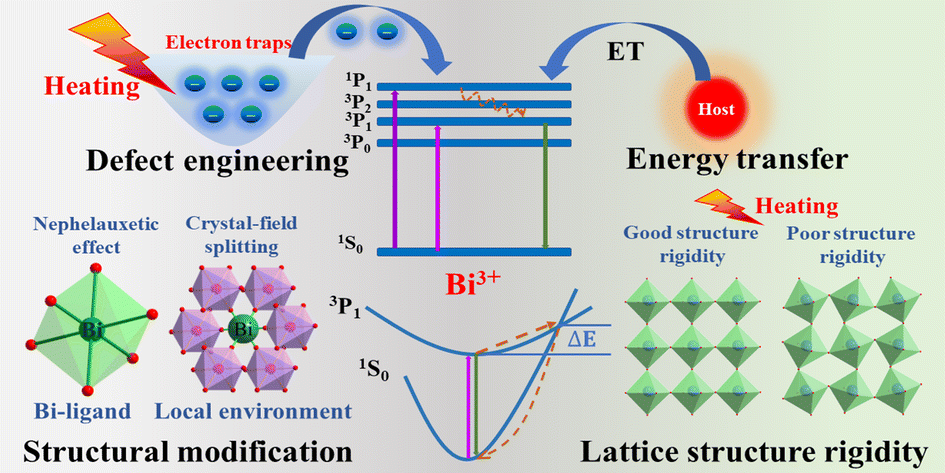
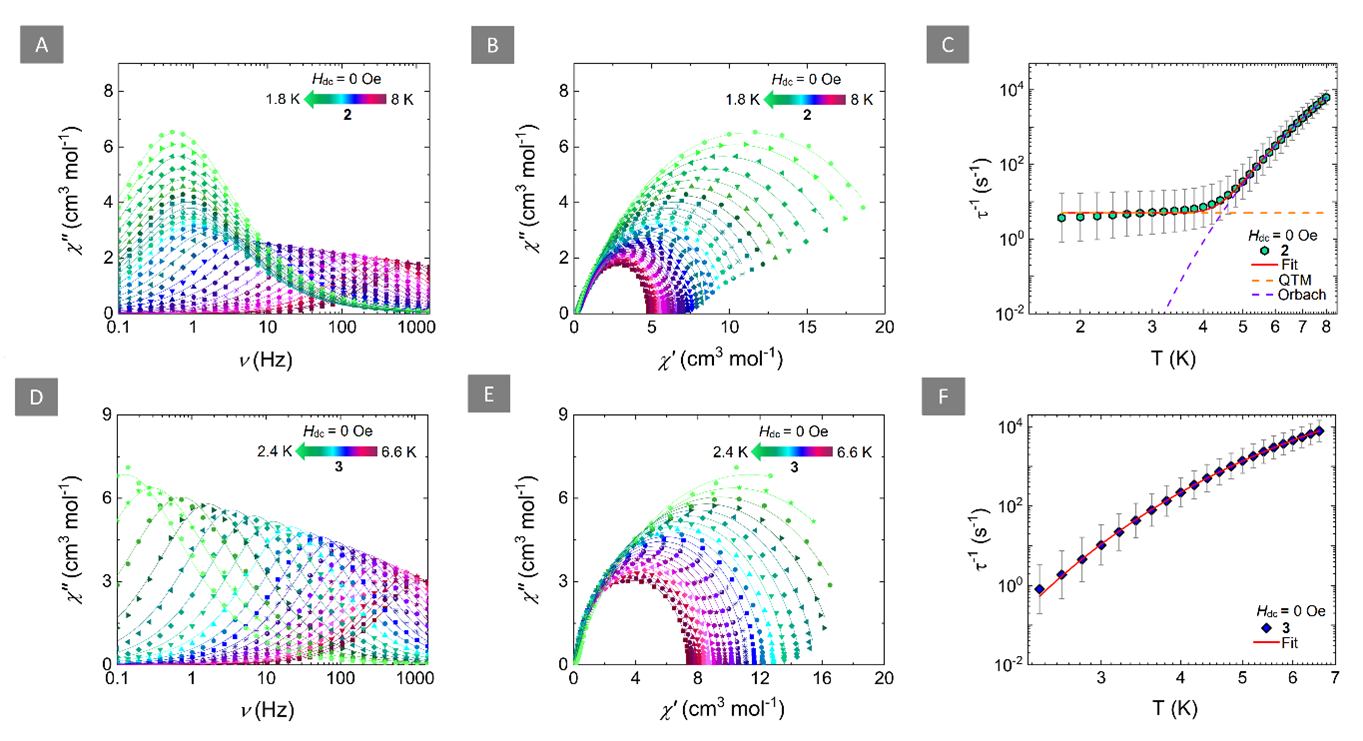
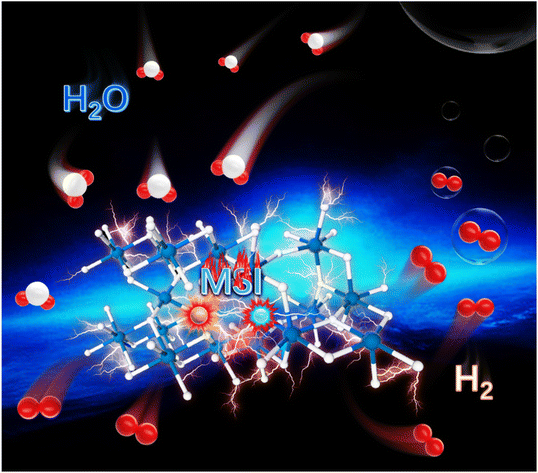
Achieving ultrasensitive temperature sensing through non-thermally coupled energy levels to overcome energy gap constraints
|
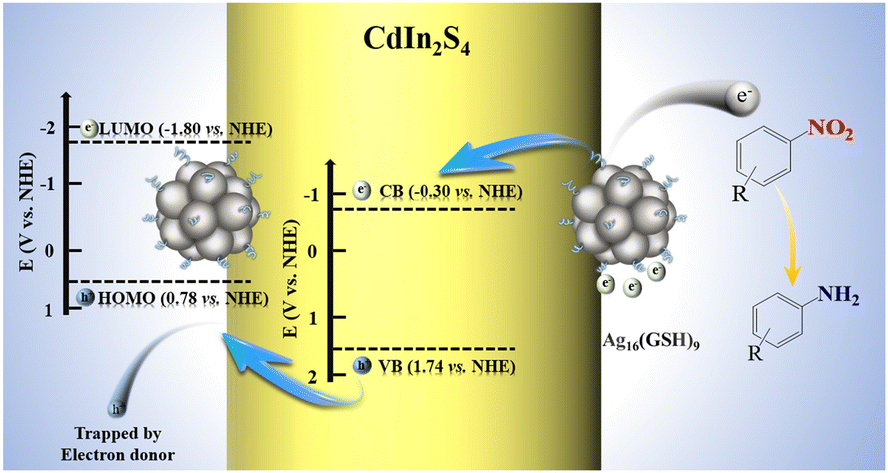 |
Photoredox catalysis enabled by atomically precise metal nanoclusters
Junyi Zhang, Linjian Zhan, Boyuan Ning, Yunhui He, Guangcan Xiao, Zhixin Chen and Fang-Xing Xiao |
 |
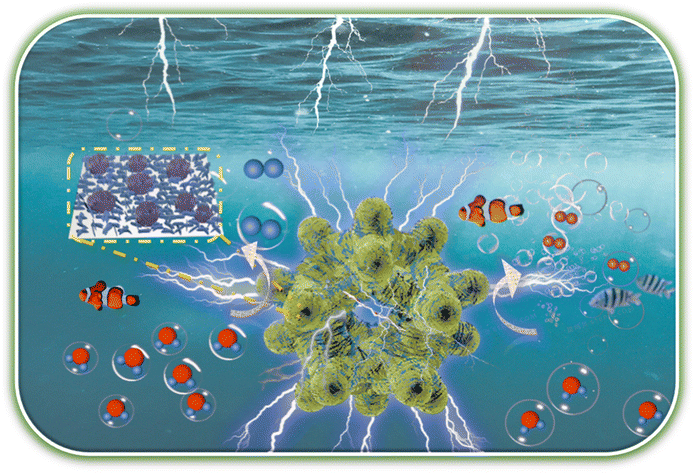
Low-content Ru–Pt supported on oxygen vacancy enriched black TiO2 with strong electronic interactions as efficient hydrogen generation electrocatalysts
Yuanzong Shen, Weichen Li, Wenna Wang, Liantao Xin, Weiping Xiao, Guangrui Xu, Dehong Chen,d Lei Wang, Fusheng Liu and Zexing Wu |
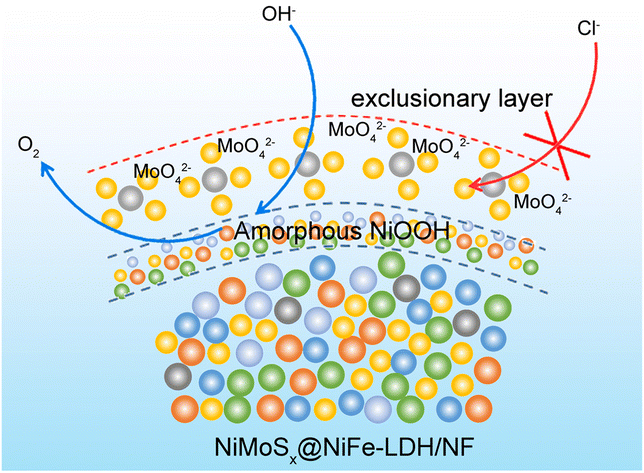 |
Highly efficient and stable oxygen evolution from seawater enabled by a hierarchical NiMoSx microcolumn@NiFe-layered double hydroxide nanosheet array
|
 |
Hierarchical porous NiFe-P@NC as an efficient electrocatalyst for alkaline hydrogen production and seawater electrolysis at high current density
Zhi Chen, Qichang Li, Huimin Xiang, Yue Wang, Pengfei Yang, Chunlong Dai, Huadong Zhang, Weiping Xiao, Zexing Wu and Lei Wang |
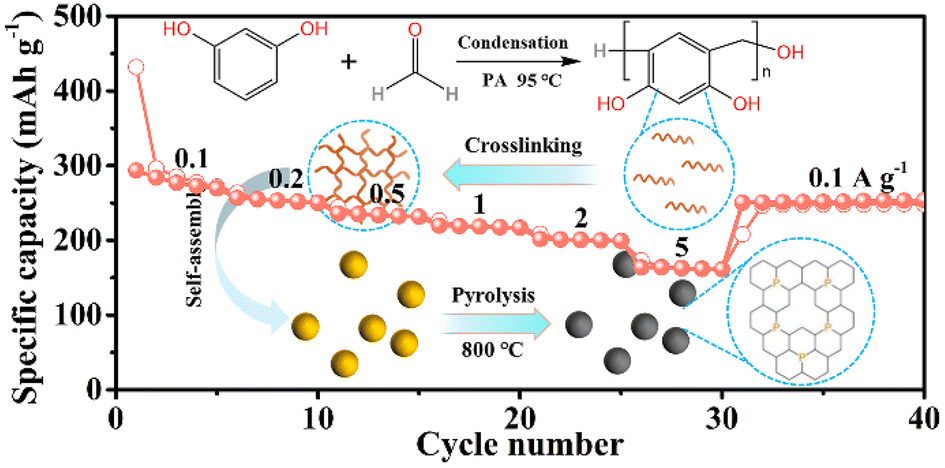 |
P-doped hard carbon microspheres for sodium-ion battery anodes with superior rate and cyclic performance
|
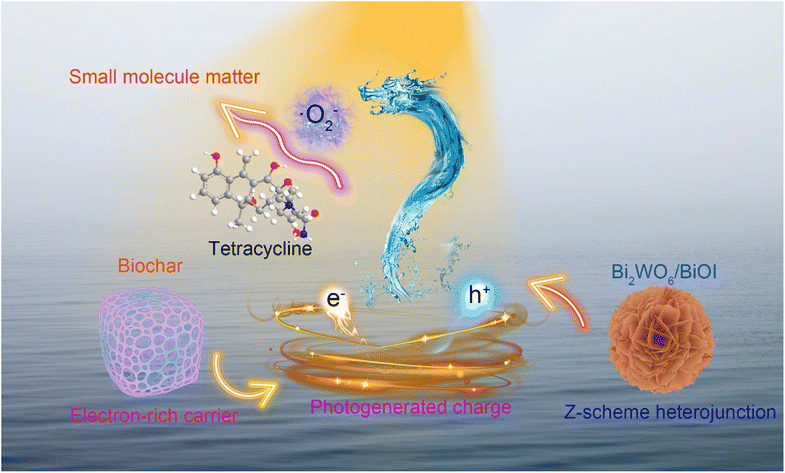 |
Electron-rich biochar enhanced Z-scheme heterojunctioned bismuth tungstate/bismuth oxyiodide removing tetracycline
Fuyan Kang, Xiaona Jiang, Yao Wang, Juanna Ren, Ben Bin Xu, Guoyang Gao, Zhanhua Huang and Zhanhu Guo |
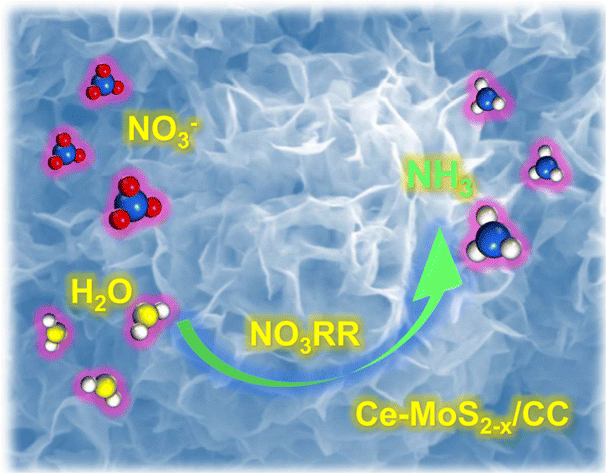 |
Ce-doped MoS2−x nanoflower arrays for electrocatalytic nitrate reduction to ammonia
Yaojing Luo, Kai Chen, Guohui Wang, Guike Zhang, Nana Zhang and Ke Chu |








")