Amino acids form the fundamental building blocks of proteins and peptides and largely control the biochemical and biophysical properties of a living organism. Incorporation of fluorine in amino acids has been an area of wide interest, because replacement of a hydrogen atom with a fluorine atom leads to a significant change in the electronic properties of a substrate and could potentially lead to a wide variation in its biological activity. The most common functional group introduced in this regard is the trifluoromethyl (-CF3) group, which provides an alternative to the -CH3 group in terms of its electronic properties. One very commonly available source of the -CF3 group is ethyl 3,3,3-trifluoromethyl pyruvate, which was utilized very elegantly by Professor Mario Waser and his group in their recent report published from the Johannes Kepler University, Linz to form α-trifluoromethylated proline derivatives, which are potentially very interesting surrogates for naturally occurring proline.
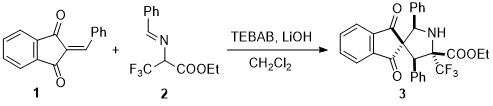
Scheme 1: Diastereoselective formal (3+2) cyclization to form α-CF3 proline derivatives
Benzyl imine derived from ethyl 3,3,3-trifluoromethyl pyruvate was treated with benzylidene indanedione (1) in the presence of LiOH to form the spirocyclic α-trifluoromethyl proline derivative 3 as the sole diastereomer (Scheme 1). It was observed that simple ammonium salts used as phase transfer catalysts could improve the conversion drastically and benzyl triethylammonium bromide (TEBAB) was found to be the best reagent for the same. This transformation exhibits a wide substrate scope with different acceptors and donors alike, while maintaining very good diastereoselectivities.
Mechanistically, the transformation is driven by the formation of the stable 2-azaallyl carbanions (4 and 4’), which by virtue of its two resonating forms could form the two different spirocyclic regioisomers 3 and 5 (Scheme 2). It was observed that the α-nucleophilic attack on the Michael acceptor 1 proceeded exclusively and there was no trace of the γ-adduct for any of the substrates. The remarkable levels of diastereoselectivity of the α-adducts adds to the ingenuity of the method.
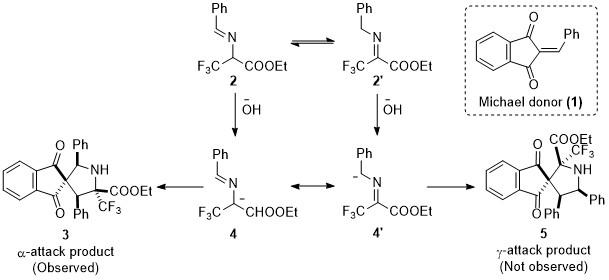
Scheme 2: Formal (3+2) cyclization modes leading to α-CF3 proline derivatives
This is indeed a very useful and efficient method for the construction of an unprecedented class of α-trifluoromethylated proline derivatives which can easily be incorporated in peptide chains for biological studies. There is however, room for improvement in this methodology, as the efforts towards enantioselective formation of the trifluoromethylated proline derivatives were unfortunately met with failures. The authors used cinchona alkaloid derived chiral phase transfer catalysts, which resulted in unsatisfactory enantioselectivities. A successful enantioselective protocol towards the formation of fluorinated amino acids would drive this field of research even further in future.
About the blog writer: Satrajit Indu is a recent PhD graduate from Indian Institute of Technology Bombay. His doctoral work in the group of Prof. Krishna P. Kaliappan was mainly focused on the total synthesis of complex natural products and development of new catalytic methods aimed for achieving interesting chemical transformations. A keen interest in organic chemistry coupled with an urge to communicate with the scientific community has driven him to take up blog writing.








")




{kind=link}