Hole-mediated PhotoRedox Catalysis: Tris(p-substituted)biarylaminium Radical Cations as Tunable, Precomplexing and Potent Photooxidants.
Processes involving visible light photoinduced electron transfer (PET) are at the forefront of contemporary organic synthesis and allow access to reactive intermediates unavailable from conventional chemical reactivity. The selective delivery of photon energy to visible light-active photocatalysts which engage visible light-inactive molecules in PET is now an established synthetic technology known as PhotoRedox Catalysis (PRC). PRC is broadly-applicable, selective, proceeds under exceedingly mild conditions and is changing the way we do organic synthesis. However, PRC suffers some key limitations. Firstly, excess sacrificial chemical oxidants (O2) or reductants (trialkylamines) are needed to turn over the ‘spent’ photocatalyst which can (or whose by-products, such as peroxides, can) interfere with subsequent chemical processes and need separation from desired products. Secondly, the scope of redox processes is fundamentally restricted by the energy of visible light photons (400-700 nm; 1.8-3.1 eV) and not all of this photon energy provided to the photocatalyst can be harnessed synthetically. Although high energy visible photons (400-450 nm) can temporarily populate excited states higher than the first excited state, Kasha’s rule dictates that relaxation to the first excited state is faster than typical diffusion-controlled photochemistry. This limits the applications of PRC in challenging redox processes.
Nature overcomes photon energy limitations by accumulating multiple photons for challenging chemical processes; several red photons are required for transfer of electrons to CO2 in biological photosynthesis. With some exceptions, harnessing of multiple photon energies in PRC to access super-oxidizing or super-reducing excited states has largely eluded researchers. The fusion of photochemistry and electrochemistry provides an innovative solution to all issues above. Electroactivation of a catalyst to a colored radical ion, followed by its photoexcitation, exceeds the accessible excited state redox potentials of PRC alone (>3.0 eV). Some examples of this concept, coined ‘electrochemically-mediated PhotoRedox Catalysis’ (e-PRC), have emerged in the literature. A question that has eluded researchers is how excited radical ions (typically doublet states) could ever engage in photochemistry, given their typical picosecond lifetimes forbid diffusion. Moreover, while catalyst ‘tunability’ is well established in PRC, it is yet to be established in synthetic photoelectrochemistry.
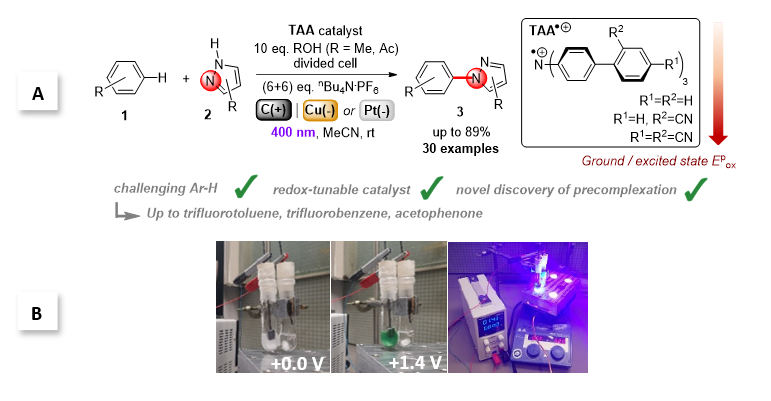
Figure 1. Triarylamines (TAAs) as tunable e-PRCats developed by the Barham grou.; A. SET activation of challenging arenes and C-N bond formation with N-heterocycles. B. Experimental setup showing electroactivation of TAAs and photoexcitation of TAA.+s in a divided cell.
Recently, the group of Joshua Philip Barham and collaborators at Universität Regensburg, Technische Universität München and the Central European Institute of Technology introduced triarylamines (TAA) as a new family of tunable electroactivated photoredox catalyst (e-PRCat). The group demonstrated that facile tuning of the e-PRCat accessed record-breaking excited state potentials of Epox = +4.4 V vs SCE. This allowed PET super-oxidations of very challenging arenes, like polychloroarenes, polyfluoroarenes and trifluorotoluene, resulting overall in C-N bond formation with pyrazole partners. Catalyst power could be tuned down to access moderately challenging arenes (alkylbenzenes, benzene) with higher selectivity. Of key importance is the discovery of p-stacking dispersion precomplexation between the radical ion e-PRCat (TAA.+) and substrate to rationalize, for the first time, the photochemistry of excited state radical ions. The study sets the scene for dispersive precomplexation as a novel control element in photochemistry, which allowed the group to:
1) circumvent of ultrashort lifetimes of radical ion excited states (*TAA.+, t < 10 ps) for use in PET,
2) override Kasha’s rule and access to higher order excited states, to harness the full power of the visible photon’s energy (Epox *TAA.+ > 4.0 V vs. SCE),
3) overturn conventional thermodynamic redox selectivity (1,4- > 1,2-disubstituted arenes) by steric/electronic factors involved in precomplexation (1,2- > 1,4-disubstituted arenes).
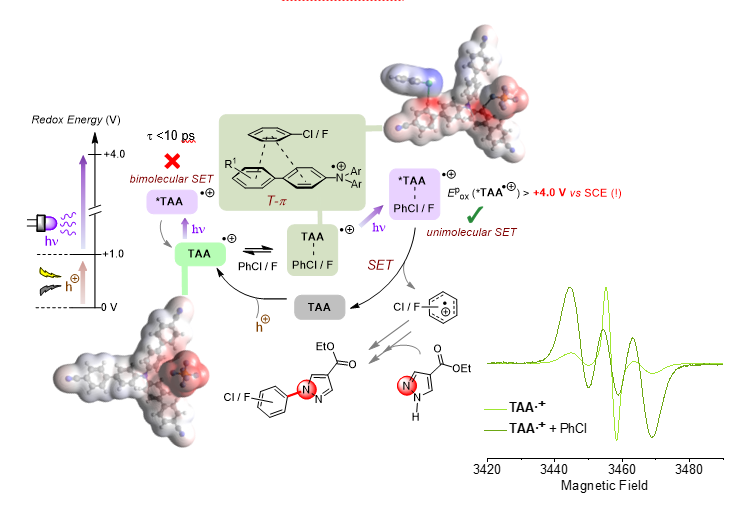
Figure 2. Proposed mechanism of hole-mediated photoredox catalytic super-oxidation of arenes, involving dispersive precomplexation via a T-p interaction, supported by DFT calculations and changes in EPR spectroscopy of the TAA.+ in the presence of arene substrates following a shift in spin density (increasing triplet representation of the signal).

Joshua Philip Barham
Universität Regensburg
Joshua Philip Barham is a Sofja Kovalevskaja Group Leader in the Faculty of Chemistry and Pharmacy at the University of Regensburg (Germany), where he investigates photo-, electro-, photoelectro- and flow chemistry as enabling technologies in organic synthesis. He received his industry-based Ph.D in Chemistry in 2017 under the supervision of Prof. John Murphy at the University of Strathclyde (U.K.) and Dr. Matthew John at GlaxoSmithKline (U.K.). His postdoctoral studies with Prof. Yasuo Norikane and Prof. Yoshitaka Hamashima at the National Institute of Advanced Industrial Science and Technology and the University of Shizuoka (Japan) specialized in photoredox catalysis and microwave flow chemistry. In addition to his authorship of 20 articles indexed by SCI which have been cited ~450 times, he has authored a book chapter, a patent, an industrial press release and various blogs/webinars. For a complete list of publications, see: http://www-oc.chemie.uni-regensburg.de/barham/page_417_en.php
https://scholar.google.co.uk/citations?user=fBgXhboAAAAJ&hl=en








")