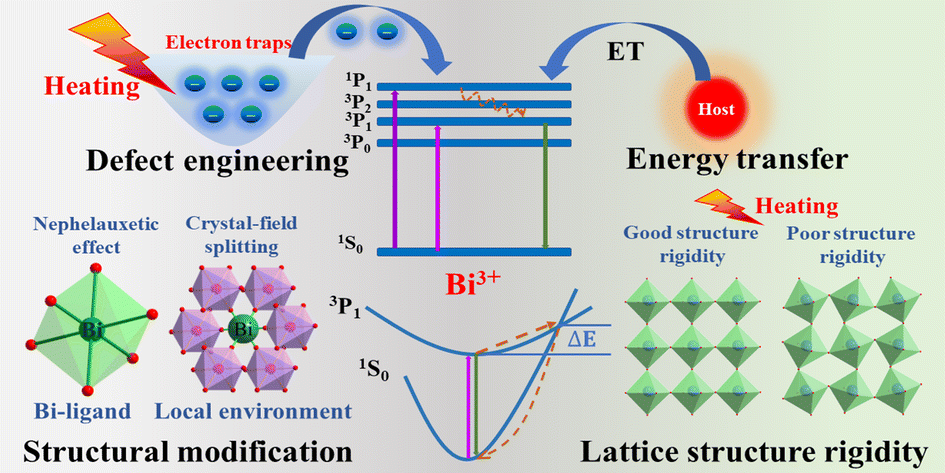
From unprecedented 2,2′-bisimidazole-bridged rare earth organometallics to magnetic hysteresis in the dysprosium congener
Florian Benner and Selvan Demir
Inorg. Chem. Front., 2023, 10, 4981-4992
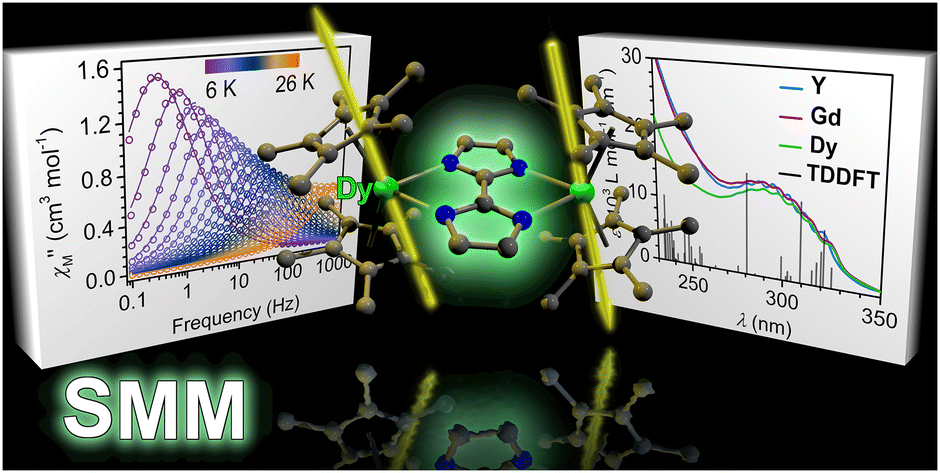
Single-molecule magnets (SMMs) are molecules that show slow magnetic relaxation, originating from a bistable magnetic ground state with a thermal barrier to spin relaxation (Ueff). Remarkably, SMM can exhibit open magnetic hysteresis loops which correspond to retaining magnetic memory just like tiny bar magnets can. This property renders SMMs exciting for potential applications in high-density information storage, magnetic refrigeration, quantum computing and spintronics. Over the last years, the SMM field exploited mononuclear dysprosium metallocenium cations as spin carriers, where the well-defined coordination sphere imposed by cyclopentadienyl ligands strongly amplifies the easy axis of the dysprosium(III) ion. To date, synthetically accessible single-ion magnets operate at best slightly above the boiling temperature of liquid nitrogen (77 K). Consequently, it was realized that lanthanide ions must be strongly coupled to one another to increase operating temperatures, ideally towards room temperature. To this end, the nature of the bridging ligand is vital and the exploration of new organic bridging ligands along with their utility in coupling lanthanide metallocene fragments is crucial. That knowledge will aid to devise design principles of SMMs with amplified magnetic coupling between lanthanide metallocene moieties.
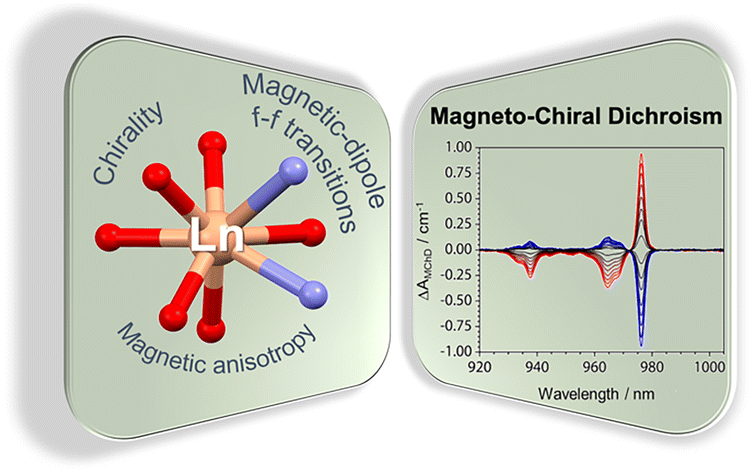
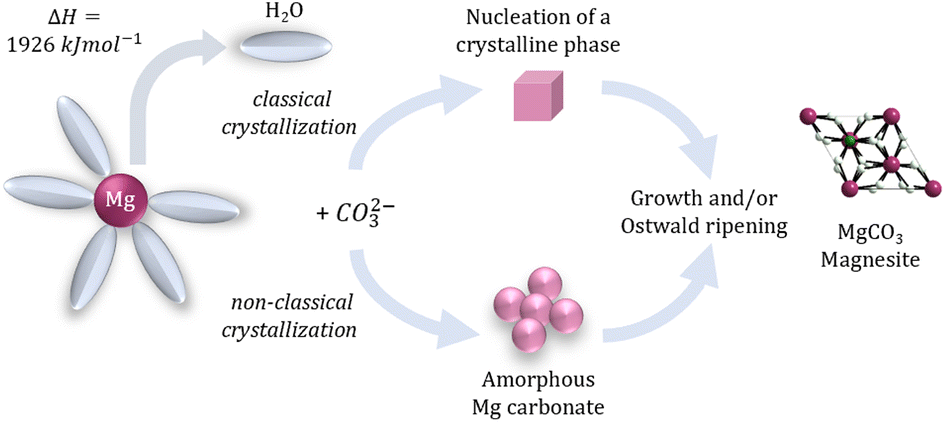
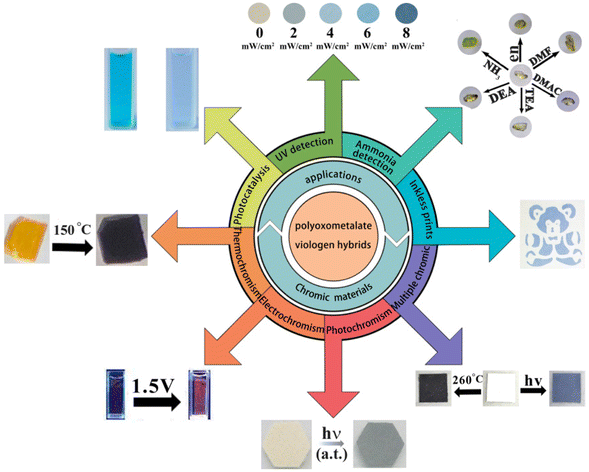
Recently, the group of Selvan Demir at Michigan State University implemented the bridging ligand 2,2′-bisimidazole for the first time into rare earth and magnetochemistry, where this tetranitrogen ligand connects two metallocenium units (Figure 1). The synthesized series consists of three compounds comprising the diamagnetic yttrium, the paramagnetic gadolinium (isotropic) and paramagnetic dysprosium (anisotropic) ions. Excitingly, the dinuclear dysprosium complex features SMM behavior and on the timescale of magnetic hysteresis measurements, open hysteresis loops of up to 5 K. The half-filled f-electron valence shell for trivalent gadolinium ions allows quantification of the magnetic exchange coupling since the orbital singlet affords magnetic behavior that is free of complications arising from spin-orbit coupling. Thus, dc magnetic susceptibility measurements on the respective gadolinium complex revealed weak antiferromagnetic interaction between the metal ions. Due to its comparable ionic size, the yttrium analog served as a diamagnetic surrogate to the lanthanides, enabling the in-depth investigation of the electronic structure of these complexes via spectroscopic methods and density functional theory (DFT) calculations. In this way, absorption spectra were related to the underlying electronic structure of these complexes, revealing prevalent excitations from the predominantly ligand-based frontier orbitals into metal-based higher-lying orbitals. This provided also profound insight into the redox (in)activity of the bisimidazole bridge, which, in contrast to its annulated 2,2′-bisbenzimidazole analog, showed no reactivity towards reductants or oxidants. This was primarily ascribed to the title compounds lacking accessible ligand-based low-lying π* orbitals, unlike the opposite observation for the respective bisbenzimidazole counterparts. In sum, the fact that the bisimidazole ligand retains and enhances the single-ion anisotropy of the dysprosium ions while providing a wealth of substitution sites for future chemical modification renders this ligand system highly promising for the construction of higher nuclearity systems.
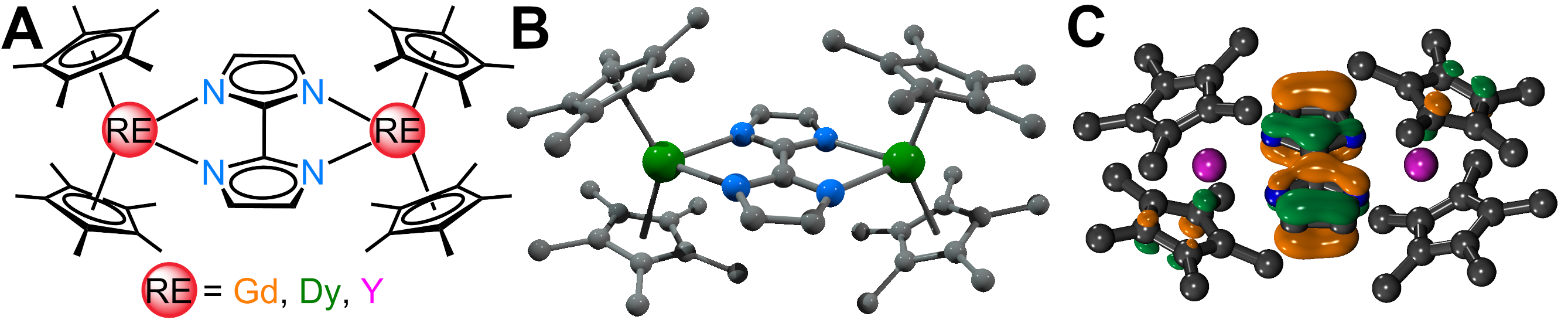
Figure 1. A: Schematic view of the bisimidazole-bridged rare earth metallocene complexes. B: Structure of the complexes as determined through single-crystal X-ray diffraction analysis. C: Plot of the bisimidazole-centered highest occupied molecular orbital of the yttrium complex.
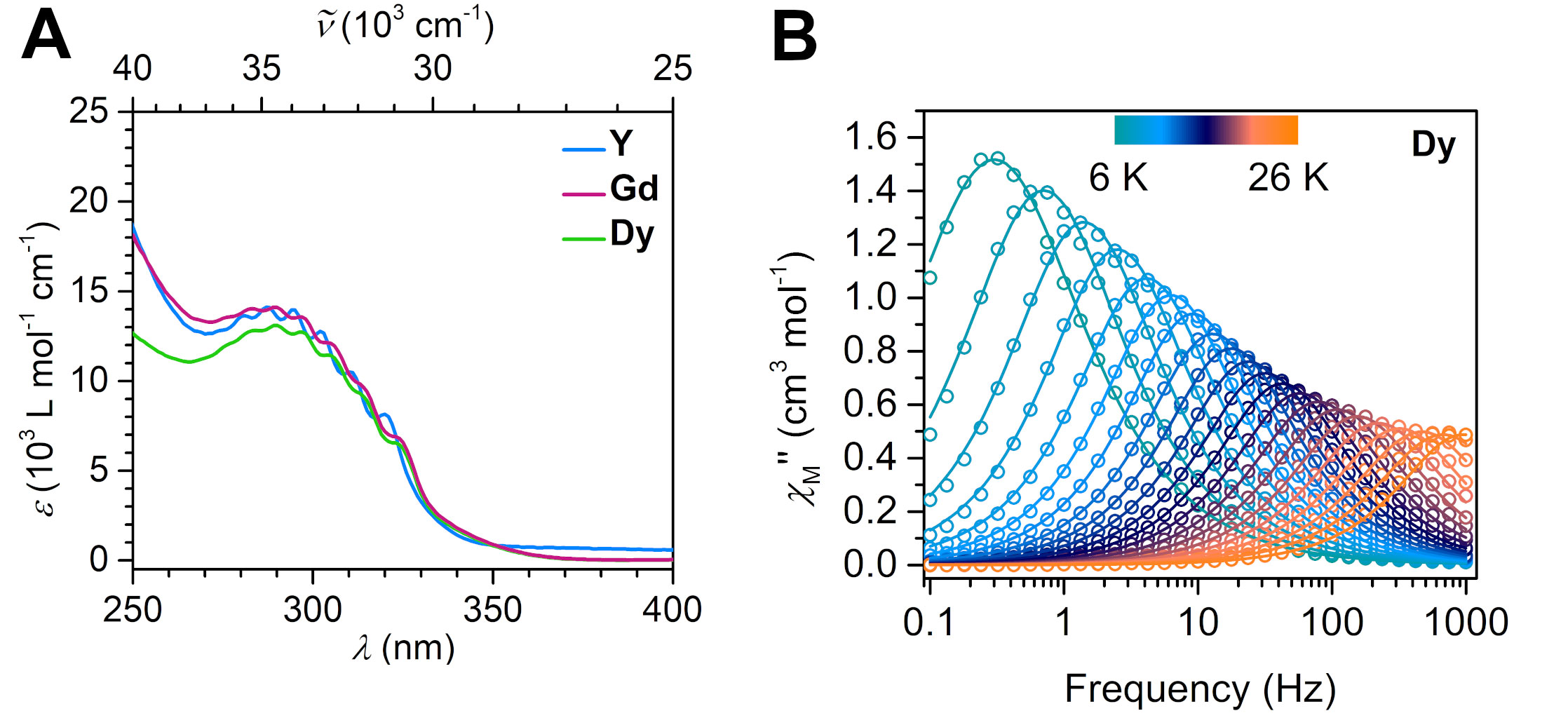
Figure 2. A: Absorption spectra of the rare earth complexes in the ultraviolet/visible region of the electromagnetic spectrum. B: Dynamic magnetic measurement revealed slow magnetic relaxation and single-molecule magnet behavior for the dysprosium complex.
Corresponding Author:

Selvan Demir is an Assistant Professor of Chemistry at Michigan State University. She received a Dr. rer. nat. in Chemistry from the University of Cologne researching on scandium solid state chemistry with Prof. Gerd Meyer and scandium organometallic chemistry with Prof. William J. Evans at the University of California, Irvine. Subsequently, she was a Postdoctoral Scholar, where she conducted research on lanthanide-based single-molecule magnets and porous aromatic frameworks with Prof. Jeffrey R. Long at the University of California, Berkeley. Simultaneously, she explored the transuranics with Dr. David K. Shuh at the Lawrence Berkeley National Laboratory. Afterwards, she took up a junior professorship of inorganic chemistry at the University of Göttingen. Since 2019, she researches with her group at Michigan State University, on various areas surrounding the chemistry of the rare earth elements and actinides. Her research program has a strong emphasis on organometallic chemistry, small molecule activation, organic radicals, single-molecule magnets, qubits, bismuth chemistry, dibenzocyclooctatetraene chemistry, and lanthanide/actinide separations.
Comments Off on Bisimidazole – Exciting for Organometallics and Single-Molecule Magnets








")